Online Structure Alignment Resources (April 2005;
later additions as noted)
This list is not meant to be comprehensive, but includes a
reasonably broad sample of sites I think are handy and convenient to use.
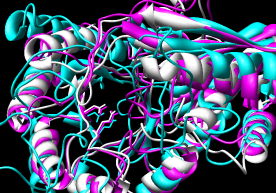
The test cases involve structures with low pairwise sequence identity:
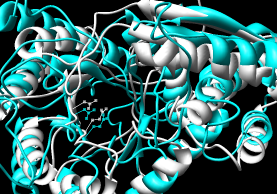
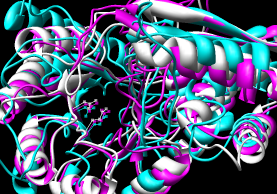
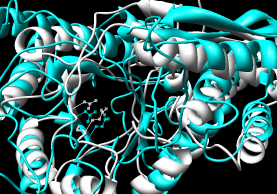
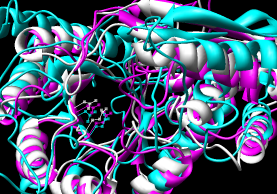
- Pairwise - 2mnr (mandelate racemase) 4enl (enolase)
- Multiple - 2mnr 4enl 1nu5 (MLE II)
Default settings generally used.
Note that the
MatchMaker tool
in Chimera performs
a similar function: it constructs a sequence alignment
and superimposes the structures accordingly.
Secondary structure can be used
to help construct the alignment, which allows pairs with lower
percent identity to be superimposed correctly
(see
results from using MatchMaker).
Superposition Servers
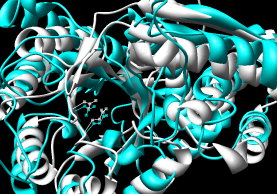
- FAST
- pairwise; paper compares the method to DaliLite, CE, and K2.
Reference:
Zhu J, Weng Z.
FAST: a novel protein structure alignment algorithm.
Proteins. 2005 Feb 15;58(3):618-27.
Availability:
server;
executables for Linux, IBM, Sun, Alpha, Mac OS X can be downloaded
Performance:
~5 seconds for pairwise.
Shows a sequence alignment in the web page;
superimposed coordinates are available within the downloadable
RasMol script (puts both into chains into one model and makes them
A and B; Chimera complains about the bad PDB records
but will display the structures nonetheless).
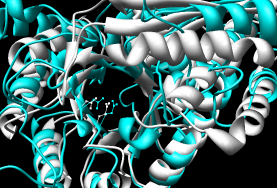
- MultiProt
- truly multiple rather than assembly of pairwise alignments;
can handle circular permutation.
Method seems elaborate (opinion from reading the paper) but works well.
Reference:
Shatsky M, Nussinov R, Wolfson HJ.
A method for simultaneous alignment of multiple protein structures.
Proteins. 2004 Jul 1;56(1):143-56.
Availability:
server;
can be downloaded for Linux
Performance: ~28 seconds for 3-way using sequence order.
Reports several alignments: five of all three, five with various pairings.
I took the 3-way alignment with the most residues aligned.
Coordinates are combined in a single file but come out as separate
submodels in Chimera (#0.1, #0.2, #0.3).
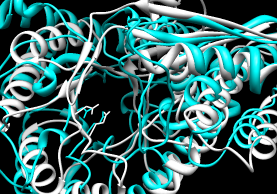
- SuperPose
- multiple; authors claim the server is especially user-friendly.
Reference:
Maiti R, Van Domselaar GH, Zhang H, Wishart DS.
SuperPose: a simple server for sophisticated structural superposition.
Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W590-4.
Availability:
server
(uses a number of other programs including VADAR and ClustalW)
Performance:
~25 seconds for pairwise
(not including waiting for the display applet).
Results page allows download of superposition in PDB format,
sequence alignment, and
RMSD information.
Coordinates are combined in a single file but come out as separate
submodels in Chimera (#0.1, #0.2, #0.3).
Only two files can be uploaded, so according to the instructions you have to
combine input files to superimpose more than two structures.
I had trouble getting this to work. The support person said
the structures must have chain IDs; however, adding chain IDs to
my input did not work. Only taking the advice to use the server to
superimpose two and then using that output (which, incidentally,
did NOT include chain IDs) as one of the inputs for
another round of superposition worked.
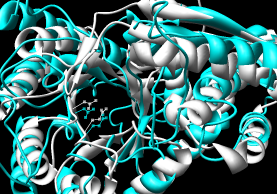
- SALIGN (updated June 2012) - multiple
Reference:
Braberg H, Webb B, Tjioe E, Pieper U, Sali A, Madhsudhan MS.
SALIGN: A Webserver for alignment of multiple protein sequences and structures.
Bioinformatics. 2012 May 21. [Epub ahead of print]
Availability:
server
or
DBAli lookup
Performance:
Notification of server calculations is by email generally within an hour,
depending on the server load, and the results page includes a link to
“launch Chimera” to display the superimposed structures and
sequence alignment. DBAli database lookup takes a few seconds and
the resulting web page shows the superposition in a Jmol window
and a sequence alignment. The sequence alignment can be downloaded
in PIR, FASTA, or PAP format, and a
PDB file of the superimposed coordinates
can be downloaded.
Coordinates are combined in a single file but come out as separate
submodels in Chimera (#0.1, #0.2, #0.3).
These were not in the order of specification in the
input file, but the submodels
were also given chain IDs (A, B, C) that did reflect that order.
Rather than using that PDB file,
I find it easier to open the alignment in Chimera, autoload
the structures from the PDB (since the sequences are named with
the PDB IDs), and match the structures using the sequences,
because this retains the original chain IDs plus ligands, ions, etc.
|
- K2, K2SA
- pairwise; the program (but not the server?)
can handle circular permutation.
Reference:
Szustakowski JD and Weng Z.
Protein structure alignment using a genetic algorithm.
Proteins, 38(4):428-440, Mar 2000.
Availability:
server
Performance: ~30 seconds using sequential constraints.
Can download coordinates for second protein (transformed to
match first protein) and a log file.
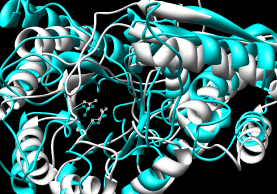
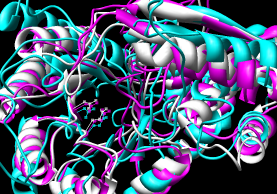
- CE (pairwise), CE-MC (multiple, building upon
pairwise CE alignments)
 |
 |
| CE |
CE-MC |
References:
Shindyalov IN, Bourne PE.
Protein structure alignment by incremental combinatorial extension (CE)
of the optimal path.
Protein Eng. 1998 Sep;11(9):739-47.
Guda C, Lu S, Scheeff ED, Bourne PE, Shindyalov IN.
CE-MC: a multiple protein structure alignment server.
Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W100-3.
Availability:
CE and CE-MC servers used in test no longer available, but see
jCE server
and software download
Performance:
~55 seconds for pairwise (CE server). ~4 minutes for multiple alignment
(CE-MC); e-mail address required for this option.
Results include structure-based sequence alignment and
single downloadable PDB in which the structures
are given different chain IDs (Chimera complains about duplicate
atom serial numbers but displays the structures nonetheless).
In the CE-MC results, the residue numbers are shifted over one column
from where they should be, so the file has to be edited before viewing.
The support person said the problem would be fixed in the next update.
- DaliLite - pairwise
(the Dali server instead
compares a single input structure against a representative set from the PDB,
like VAST Search)
Reference:
Holm L, Park J.
DaliLite workbench for protein structure comparison.
Bioinformatics. 2000 Jun;16(6):566-7.
Availability:
server used in test no longer available, but see newer-version
DaliLite server
Performance: ~1 minute; reports several alignments and their stats;
for each, coordinates can be downloaded for the second protein.
The figure shows the superposition with the most residues aligned.
- Protein3Dfit (added July 2005) - pairwise
Reference:
Lessel U, Schomburg D.
Similarities between protein 3-D structures.
Protein Engineering. 1994 Oct;7(10):1175-87.
Availability:
server (URL updated Feb 2009)
Performance:
~25 seconds for pairwise (default settings; various parameters
can be adjusted by the user).
The resulting web page shows the superposition in a
Jmol window plus a sequence alignment and RMSD table. Links include
many different ways (convenient!) to download the results.
One way to save the superposition is by downloading the
transformed coordinates of the second structure, and
the sequence alignment can be saved in
PIR format.
- CLICK (added Oct 2011) - pairwise,
sequence-order-independent, multiple solutions returned
Reference:
Nguyen MN, Tan KP, Madhusudhan MS.
CLICK--topology-independent comparison of biomolecular 3D structures.
Nucleic Acids Res. 2011 Jul;39(Web Server issue):W24-8.
Availability: server
Performance:
~35 seconds for pairwise (default settings; parameters can be adjusted).
The resulting web page shows solutions in order of decreasing % structure
overlap, each with a Jmol window and (fragmentary) sequence alignment.
Results can be downloaded as PDB files of the superimposed structures.
- later additions, not evaluated:
- ARTS
(for nucleic acid structures)
- FATCAT (Flexible structure AlignmenT by Chaining Aligned
fragment pairs allowing Twists; i.e., one of the
structures may be split into pieces)
- POSA
(flexible; multiple; e-mail address requested but not mandatory)
- RCSB PDB protein comparison tool
(pairwise comparison with java-FATCAT, java-CE, FATCAT, Mammoth,
TM-Align, TopMatch, or sequence alignment methods)
- GANGSTA+
(seq-order-independent; server options require e-mail address)
- TopMatch (successor of ProSup)
(see also TopSearch for multiprotein complexes)
- PDBj
superposition servers (RASH and GASH) - seek to maximize NER (number
of equivalent residues) at 4 Å cutoff
- C-alpha match
(pairwise, seq-order-independent)
- PDBeFold
- 3D-SS (choice of method: STAMP or ProFit)
- see also
Structural alignment software at Wikipedia
Circularly Permuted Test System (May 2005)
Test structures: 1qq5 A chain (HAD) 3chy (CheY)
These are in different superfamilies according to SCOP,
but it has been noted that the structures appear to
be related by circular permutation (see
paper by Ridder and Dijkstra).
The active site
residues are in about the same positions in 3D, although
in different N→C orders. Selected active site residues
(those in a column are aligned in 3D):
-----------------------------------------------------------
loop 1 loop 2 loop 3 loop4
-----------------------------------------------------------
2-HAD X. autrophicus *
D8 S114 K147 171SS---D176
epoxide hydrolase M. musculus, H. sapiens *
D9 T123 K160 184DD---N189
CheY E. coli, S. typhimurium *
D57 T87 K109 12DD13
-----------------------------------------------------------
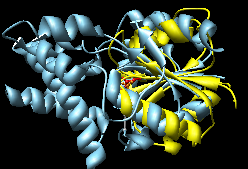
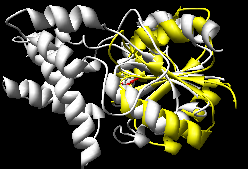
| superimposed manually |
MultiProt (~10-15 seconds)
(the choice with the most residues aligned
and as a tie-breaker, the lowest RMSD) |
K2SA (~15-20 seconds)
(wrong, see log file) |
 |
 |
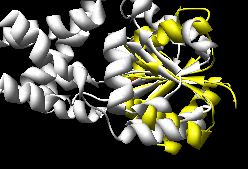 |
The Multiprot results correctly place the corresponding
active site residues together; the K2SA results are shifted
over by one strand in the central sheet.
NB: The K2SA server appears to be imposing sequential
constraints even when I set the option to NOT do so...
which would explain the failure on this test case.
Sources of Structure-Based Sequence Alignments
(which can then be used in Chimera's
Multalign Viewer
to match the corresponding structures)
Listed here are Web servers that generate alignments from your input.
Databases of precalculated alignments are listed
elsewhere.
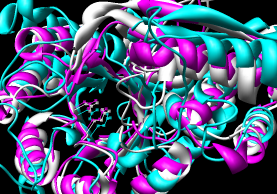
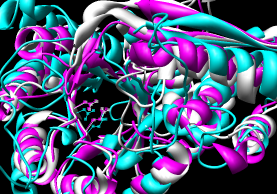
- 3DCoffee
- multiple; primarily intended to generate
sequence alignments but can use 3D structures to do so;
can also combine multiple sequence alignments
 |
 |
| Multalign Viewer matching defaults |
with iteration (pruning) |
Reference:
Poirot O, Suhre K, Abergel C, O'Toole E, Notredame C.
3DCoffee: a web server for mixing Sequences and Structures into
multiple sequence alignments.
Nucleic Acids Research 2004 Jul 1;32(Web Server issue):W37-40.
Availability:
TCoffee server (includes 3DCoffee option)
Performance:
The three PDBs were submitted using the Advanced 3DCoffee page.
Results were returned in ~4 1/2 minutes and include an
alignment (clustal format, *.aln)
and dendogram information (*.dnd).
The superpositions were generated in Chimera by using the sequence
alignment with and without pruning of CA pairs more than 2.0 A apart.
- PROMALS3D -
web server
takes input FASTA sequences and/or PDB structures,
generates multiple sequence alignments in a few formats (see my
Mar 08
journal club presentation)
meng[at]cgl.ucsf.edu /
home page