This tutorial focuses on using the Model Panel and handling ensembles of structures (such as those determined by NMR). Note that the Model Panel is generally useful whether or not ensembles are being viewed.
To follow along with the tutorial, you will first need to download the PDB files 1dwz-edit4.pdb and 1dwy.pdb into your working directory. These files contain NMR-determined structures of a bovine prion protein fragment. 1dwz-edit4.pdb contains the first four members of an ensemble of 20 structures (present in the original PDB entry 1DWZ), whereas 1dwy.pdb is a single representative minimized structure.
On UNIX, start Chimera from the system prompt:
unix: chimeraOn Windows, click the chimera icon.
A basic Chimera window should appear after a few seconds. Make the window a convenient size, then choose the menu item File... Open. In the resulting dialog, make sure that the File type is set to PDB. Find and click 1dwz-edit4.pdb and then the Apply button so that the dialog does not disappear; next, open 1dwy.pdb, using OK to also dismiss the dialog.
Rotate, translate, and scale the structures as needed to get a better look (see mouse manipulation to review how this is done). If you like, open the Side View; choosing Tools... Viewing Parameters... Side View is one of many possible ways to do this. Continue moving and scaling the structures as desired throughout the tutorial.
Thicken the lines: Actions... Atoms/Bonds... wire width... 3. The structures include all atoms, even the hydrogens. Simplify the display using Actions... Atoms/Bonds... backbone only. Now only the N, CA, and C atoms are shown.
Open the Model Panel; choosing Tools... Inspectors... Model Panel is one of many possible ways to do this.
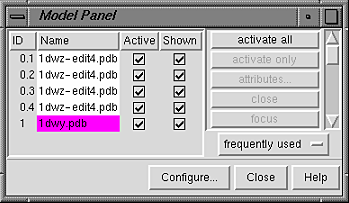
| Model Panel - initial appearance |
|---|
|
Each file of coordinates opened in Chimera becomes a model with an associated model ID number and model-level color. Some PDB files are further subdivided into multiple structures designated with MODEL and ENDMDL records; when the input file contains more than one set of such records, submodel numbers are assigned sequentially starting with 1. In this case, the four ensemble members in 1dwz-edit4.pdb are submodels 1-4 of model 0. Each submodel (0.3, for example) can be treated as a separate model. Thus, "models" will be used to indicate submodels and/or models that are not subdivided into submodels.
By default, the Model Panel shows the model-level colors behind the names. Once one or more models have been chosen within the left side, any of several functions represented by buttons on the right side may be executed. At first, most buttons are grayed out since no model has been chosen in the left side of the panel. Individual models or blocks of models may be chosen (highlighted) using the left mouse button. Ctrl-click adds to an existing choice rather than replacing it. To highlight a block of models without having to hold down the mouse button, click on the first (or last) and then Shift-click on the last (or first) in the desired block.
Click on 1dwy.pdb in the left side of the Model Panel and then try various functions on the right side:
show only - hide the other modelsin the menu: Actions... Color... by element
trace chains - display the chain trace, which includes only CA atoms
show all atoms - display all atoms
select - select the entire model for further operations
Back to the Model Panel:
sequence... opens a sequence panel for the model; click-select one or a string of residues in the sequence and see how the corresponding residues of the structure become selected. Next, Close the panel and perform some action upon the new selection, such asin the menu: Actions... Atoms/Bonds... sphere
Back to the Model Panel:
attributes... opens the molecule model attributes panel; within this,Note that using the Shown checkbox is not the same as using the command display, which works on individual atoms and bonds; instead, it enables/disables the whole model's display (see display hierarchy). Checking Shown enables the display, but the display settings of individual atoms and bonds are not changed; in this example, the hydrogens are still undisplayed, as they were before the model was hidden. Toggling checkmarks in the Shown column is the same as using the hide and show buttons; toggling checkmarks in the Active column is the same as using the activate and deactivate buttons (which control whether a model can be moved). By default, these buttons are not included on the right side of the Model Panel because they are classified as infrequently used (this can be controlled using Configure...).uncheck the Shown checkbox for 1dwy.pdb
- click the Component Residue Attributes checkbox to expand the section
- set ribbon display to on
- set ribbon depiction to rounded ribbon
- set ribbon display back to off
- click Close to dismiss the panel
check the Shown checkbox for 1dwy.pdb
uncheck the Active checkbox for 1dwy.pdb - deactivate the model for motion (so it cannot be moved with the mouse)Move the four submodels of 0 so that they do not overlap with model 1 (which is deactivated and will not move). Scaling the view down with the mouse or Side View may be helpful.
check the Shown checkbox for all of the models
check the Active checkbox for 1dwy.pdb
Choose submodel #0.1 in the Model Panel, select it, and use the Actions menu to color it. Repeat the process with the three other submodels (choosing different colors), then clear the selection (Select... Clear Selection) and Close the Model Panel.
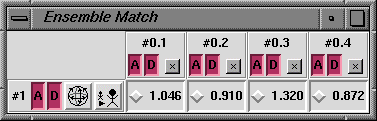
We will use the extension EnsembleMatch to compare the four NMR structures in 1dwz-edit4.pdb with the minimized structure in 1dwy.pdb. Start EnsembleMatch (Tools... Ensemble... EnsembleMatch is one way) and choose the minimized structure in model 1 as the reference and the NMR structures in model 0 as the alternative. Parts to Match are the atoms to be used for superimposing the structures. Each model contains residues 124-227 of chain A; it would be possible to use all atoms, but often it is more meaningful to compare only the backbone atoms or alpha-carbons. To use the backbone atoms N, CA, and C, enter the atom specification
@n@ca@cwhich in this case is equivalent to
:124-227.a@n@ca@cThe atom specification describes the atoms to be used in each structure. Thus, if residue numbers are given, the residues in one structure must have the same numbers as the matching residues in the other structures. If atom names are given, they should specify equal numbers of atoms occurring in the same order in the different structures (if @ca is entered, the first CA in a structure is matched with the first CA in other structures, the second CA with the second in other structures, etc.). Clicking Okay brings up the EnsembleMatch window:
 |
Finally, for fun, start EnsembleTile (Tools... Ensemble... EnsembleTile is one way). Pick model 0 as the Ensemble to Tile, and click Okay. "Tilings" are sometimes used in figures to compare different molecules or different conformations of the same molecule.
Choose File... Quit from the menu to terminate the Chimera session.