
Multalign Viewer allows viewing and manipulation of multiple sequence alignments. If at the same time the corresponding structures are displayed in Chimera, there can be crosstalk showing which regions on the structures are those highlighted in the sequences. In addition, structures can be superimposed based on the sequence alignment. For an informal introduction, see the Sequences and Structures tutorial.
MinRMS, a stand-alone program available from the UCSF Computer Graphics Lab, produces structural superpositions and other data that can be displayed with the extension AlignPlot, as well as sequence alignments that can be displayed with Multalign Viewer (and MSF Viewer). However, many other programs (beyond the Chimera suite) can generate sequence alignments suitable as input to Multalign Viewer. Likewise, structural alignments for display in Chimera can be generated in several ways other than running MinRMS, including manually.
Multalign Viewer is under development. New features will continue to be added. Currently, however, MSF Viewer may be more useful for some applications.
Multalign Viewer is an extension in the Homology category. There are several ways to start an extension. Explicitly starting Multalign Viewer brings up a dialog for opening files. File formats that can be read (and written, except for RSF) are:
| Format | Prefix | Suffix |
|---|---|---|
| Aligned FASTA | afasta: | .afasta .fasta .fa |
| Aligned NBRF/PIR | pir: | .pir |
| Clustal ALN | aln: | .aln .clustal .clustalw .clustalx |
| GCG MSF | msf: | .msf |
| GCG RSF | rsf: | .rsf |
One of these types can be chosen from the File type: pulldown menu near the bottom of the dialog, or the type can be inferred from the suffix(es) of the chosen file(s). Once a file has been read, a sequence window containing the alignment appears; if multiple files are read, each is shown in a separate sequence window. Multalign Viewer menu items are explained within the following sections, and a full listing with short descriptions is included at the end of this page.
Note that Multalign Viewer can be started automatically from AlignPlot (see the show sequence alignment feature).
| Multalign Viewer - sequence window excerpt |
|---|
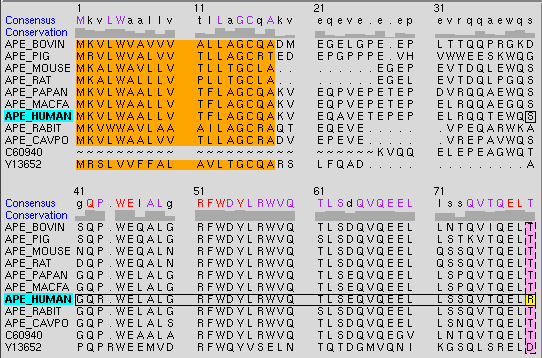
|
This figure shows part of the sequence window contents for the input file apoex.fa. A Consensus sequence and Conservation histogram are shown above the multiple alignment. In the consensus sequence, well-conserved residues (80% or greater) are capitalized and colored purple, except for the completely conserved residues, which are shown in red. Lowercase letters in the consensus sequence indicate the residue type most prevalent (not necessarily >50%) at a position in the multiple alignment. If several residue types are equally the most prevalent, one is chosen at random to appear in the consensus sequence. If a gap is most prevalent or equally the most prevalent as one or more residue types, a gap appears in the consensus sequence. The histogram bar height shows (M - 1)/(N - 1), where M is the number of occurrences of the consensus residue type at a position in the alignment and N is the number of sequences in the alignment. Thus, if every sequence has a different residue at a given position, the bar height is zero, not 1/N.
Several aspects of the sequence window appearance can be controlled in the Multalign Viewer preferences. The colored regions are described below. Tools... Find Subsequence in the Multalign Viewer menu can be used to search for the occurrence of a particular string of residues in one or all of the sequences.
When the cursor is in the sequence window, the Page Down key (or space) moves the view down to start with the block below the topmost block whose beginning is currently visible; Page Up (or Shift-space) moves the view up to start with the block above the topmost block whose beginning is currently visible.
The Hide button closes the sequence window without changing the state of Multalign Viewer. The sequence window can be reinvoked using the Raise option for the Multalign Viewer instance in the Extensions menu (abbreviated MAV). This is also useful when the window has become obscured by other windows. Help opens this manual page in a browser window, and Quit exits from Multalign Viewer.
A sequence region can be defined manually or by any of several operations within Multalign Viewer. A region is shown using a colored rectangle or outline in the sequence window; a single region can contain any number of disjoint and/or abutting rectangular blocks.
A region can be created manually by dragging with the left mouse button within the sequence window. Dragging downward into the following block highlights to the end of the preceding block. Shift-dragging with the left mouse button adds to the active region. Ctrl-dragging creates a new region and makes it the active region.
The active region is the latest region created manually, or the region last clicked on (either in the sequence window or in the listing of regions in the Region Browser). The corresponding parts of any associated structures are highlighted in the main Chimera window by becoming selected. Only one region can be active at a time. In the sequence window, the active region is indicated with a dashed outline; clicking the active region deactivates it, and clicking a different region deactivates the former active region and makes the new region active. A region with no interior color is only responsive to clicks on its borders. Where regions overlap, only the highest is responsive to clicks.
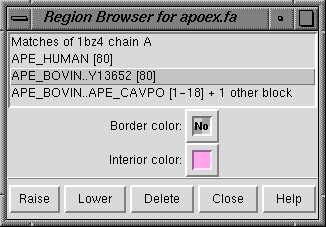 The Region Browser (Tools... Region Browser in the
Multalign Viewer menu)
controls the appearance of regions
within the sequence window.
Clicking an entry in the Region Browser makes that region
the active region;
any pre-existing active region is deactivated.
The properties shown always apply to
the active region.
Border color and Interior color
can be changed using the adjacent
color wells.
The Region Browser (Tools... Region Browser in the
Multalign Viewer menu)
controls the appearance of regions
within the sequence window.
Clicking an entry in the Region Browser makes that region
the active region;
any pre-existing active region is deactivated.
The properties shown always apply to
the active region.
Border color and Interior color
can be changed using the adjacent
color wells.
The Delete button on the Region Browser deletes the active region; the Delete key has the same effect. Since regions may overlap, a region can be considered higher or lower than another. Raise puts the active region in front of any other regions in the sequence window and moves its listing in the Region Browser to the top. Lower puts the active region behind any overlapping regions in the sequence window and moves its listing in the Region Browser to to the bottom. Alternatively, when the cursor is in the sequence window, the up arrow and down arrow keys can be used to raise and lower the active region, respectively. Cancel dismisses the Region Browser, and Help opens this manual page in a web browser.
A region newly created by dragging within the sequence window can be moved using Ctrl-left arrow and Ctrl-right arrow. Gaps can be created, extended, reduced, or removed, but residues cannot be added or removed. Flanking sequences will be pushed by moving the region until all the gap positions in the direction of movement have been exhausted.
File... Save... brings up a dialog for saving the alignment to a file. The possible formats are the same as those that can be read, with the current exception that RSF files cannot be written out.
Structures and sequences are associated automatically if certain match criteria are met: the number of gaps plus the number of residue mismatches cannot exceed 10% of the number of residues in the structure chain or the alignment sequence (whichever is shorter). The order in which the sequence and structure files are opened does not matter. Associations are reported in the status area near the bottom of the sequence window and the Chimera Reply Log. A single sequence can be associated with more than one structure. A single chain within a structure cannot be associated with more than one sequence, but different chains in the structure can be associated with different sequences. If more than one sequence matches a given structure chain, the single best-matching sequence is associated.
The names of structure-associated sequences are shown in bold over a box indicating the model-level color of the structure. The model-level color is the visible color of a structure when it is opened. For example, APE_HUMAN in the sequence window figure is associated with a structure whose model-level color is cyan. If multiple structures with different model-level colors are associated with the same sequence, the box is shown as a dark green dashed outline. When the cursor is placed over the name of a structure-associated sequence, information on the associated structure or structures is shown in the status area near the bottom of the sequence window; when the cursor is placed over a sequence residue that is associated with a residue in one or more structures, information on the structure residue(s) is given.
Sequence-structure association may create new regions. If all residues of a sequence are matched, no region is created. Otherwise, by default
Structure... Load PDBs... in the Multalign Viewer menu can be used to open PDB files corresponding to sequences that are not already structure-associated. The corresponding PDB files are determined from the sequence names using the rules given in the Structure preferences, then located and opened. If Automatically load PDBs is turned on (also in the Structure preferences), this will occur as soon as an alignment is read by Multalign Viewer.
Structure Selection by Residue Conservation
Residues within sequence-associated structures can be selected according to conservation at the corresponding positions in the sequence alignment. Structure... Select in the Multalign Viewer menu includes options to select residues at positions that that are completely conserved (100% identical), highly conserved (at least 80% identical, including the completely conserved positions), or not highly conserved (the complement of the highly conserved set).
Regions Defined by Secondary Structure
Regions can be defined within structure-associated sequences based on helices and sheets in the corresponding structures. Structure... Secondary Structure... show actual in the Multalign Viewer menu creates regions named structure helices and structure strands colored goldenrod and lime green (see the named colors), respectively. Helices and strands are defined by HELIX and SHEET records in the input file, or if these are not present, using ksdssp.
In sequences not associated with any structures, Structure... Secondary Structure... show predicted in the Multalign Viewer menu creates regions named predicted helices and predicted strands colored gold and light green (see the named colors) that correspond to helices and strands, respectively, predicted using GOR.
Structure... Match... in the Multalign Viewer menu opens a dialog with two subpanels, each listing the structures associated with sequences in the alignment. One reference structure should be chosen in the left side, but any number of structures to be matched (superimposed) with it can be chosen in the right side. The residues in each structure to be superimposed are matched to the aligned (in the sequence alignment) residues of the reference structure. If both structures are associated with the same sequence, the correspondence is even more obvious. One point per residue is used for the least-squares fitting: CA in amino acid residues and C4' in nucleic acid residues. The number of atom pairs fitted and the resulting RMSD are reported in the Chimera Reply Log and the status area near the bottom of the sequence window.
Match highly conserved residues only causes only the well-conserved (at least 80%) positions in the alignment to be used for the least-squares fit. These are the positions shown as capital letters in the consensus sequence.
Use pseudobonds to show matched atoms indicates that lines (pseudobonds) should be drawn between the matched atoms. For each matched pair of structures, a pseudobond group is created and colored uniquely (in order, the named colors dark green, dodger blue, sienna, yellow, spring green, purple, gray, and coral are used). Each group is named matches of..., where the rest of the name indicates the structures and chains that were matched. The Pseudobond Panel can be used to change the appearance of or delete the pseudobonds.
Iterate by pruning long atom pairs until no pair exceeds [x] angstroms refers to an iterative fitting procedure: in each cycle, atom pairs are removed from the match list and the remaining pairs are fitted, until no matched pair is more than x angstroms apart (x=5.0 by default). The atom pairs removed are either the 10% farthest apart of all pairs or the 50% farthest apart of all pairs exceeding the cutoff, whichever is the lesser number of pairs. The result is that the best-matching "core" regions are maximally superimposed; conformationally dissimilar regions such as flexible loops are not included in the final fit, even though they may be aligned in the sequence alignment.
OK performs the matching (superposition) and dismisses the dialog, Cancel dismisses the dialog without performing a match, and Help opens this manual page in a browser window.
Structure... Assess match... in the Multalign Viewer menu opens a dialog for evaluating how well the sequence-aligned residues of structures are superimposed in space. The dialog has two subpanels, each listing the structures associated with sequences in the alignment. One reference structure should be chosen in the left side, but any number of structures to be compared with it can be chosen in the right side. The structures to evaluate should already be superimposed on the reference structure, but the superposition can have been generated in any way, including manually (not necessarily using Structure... Match). One point per residue is used for the comparison: CA in amino acid residues and C4' in nucleic acid residues. The distance between each reference-evaluation pair of residues aligned in the sequence alignment (or corresponding to the same residue, if the reference and evaluation structures are associated with the same sequence) is measured. Using the specified Cutoff distance, a region is created in the sequence(s) associated with the evaluation structure(s):
OK performs the comparison and dismisses the dialog, whereas Apply performs the comparison without dismissing the dialog. Close dismisses the dialog, and Help opens this manual page in a browser window.
Settings... Preferences in the Multalign Viewer menu allows editing of preferences specific to Multalign Viewer. These settings are saved together with other preferences in the Chimera preferences file. Different Multalign Viewer preferences are grouped together in sections shown as index cards, Layout and Structure.
Close dismisses the Multalign Viewer preferences tool, and Help opens this manual page in a browser window.
Note that a typed-in value will not be applied until Apply, OK (which also dismisses the preferences tool), or the Enter (return) key has been pressed. Other types of settings take effect right away.
The Layout section of the Multalign Viewer preferences controls the appearance of text in the sequence window.
The Structure section of the Multalign Viewer preferences allows automatic loading of structures that go with a sequence alignment.