This tutorial describes superimposing structures, saving positions and sessions, and creating publication-quality images. See also: presets, tips on preparing images
The subjects are enzymes with shared structural features, as described in the papers:
T. Pons, D.G. Naumoff, C. Martinez-Fleites, and L. Hernandez, "Three Acidic Residues Are at the Active Site of a Beta-Propeller Architecture in Glycoside Hydrolase Families 32, 43, 62, and 68" Proteins 54:424 (2004).
F. Alberto, C. Bignon, G. Sulzenbacher, B. Henrissat, and M. Czjzek, "The Three-Dimensional Structure of Invertase (Beta-Fructosidase) from Thermatoga maritima Reveals a Bimodular Arrangement and an Evolutionary Relationship between Retaining and Inverting Glycosidases" J Biol Chem 279:18903 (2004).
| [images shown at 25%; click for full size] |
|
|
|
|
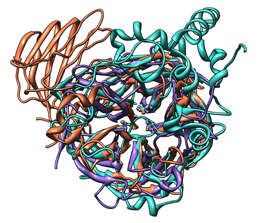
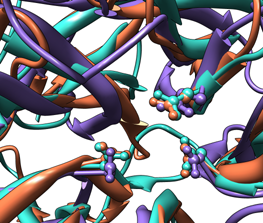
| Comparison of glycoside hydrolases from different families. Invertase (family 32) is coral, arabinanase A (family 43) is purple, and levansucrase (family 68) is turquoise. Despite differences in sequence, substrate, and catalytic mechanism, the enzymes have similar overall folds (top) and a set of highly conserved acidic residues in the active site (bottom). |
The same structures are used in the Alignments tutorial.
Glycoside hydrolases (GH) are a large, heterogeneous set of enzymes that hydrolyze bonds between sugars and other groups. Based on sequence comparisons, these enzymes have been categorized into many families; see the CAZy web site for details.
This tutorial describes the creation of images to show structural similarities among certain members of GH families 32, 43, and 68. These enzymes were grouped into different families because their relationships were not clear from sequence information alone.
The approach described on this page is only one of many possibilities. Another is to generate shadowed renderings with neon or POV-Ray.
On Windows/Mac, click the chimera icon. On UNIX, start Chimera from the system prompt:
unix: chimera
A basic Chimera window should appear after a few seconds; resize it as desired. Open the Command Line (choosing Tools... General Controls... Command Line is one way).
If you have internet connectivity, structures can be obtained directly from the Protein Data Bank. Choose File... Fetch by ID from the Chimera menu. In the resulting dialog, check the PDB option (if it is not already checked) and the option to Keep dialog up after Fetch. Fetch the PDB structures 1uyp, 1gyd, and 1oyg, in that order, and then Close the dialog. If you do not have internet connectivity, you can download the files 1uyp.pdb, 1gyd.pdb, and 1oyg.pdb into your working directory and then open them in that order as local files (with File... Open).
Move and scale the structures with the mouse in the graphics window (and optionally, the Side View) as desired throughout the tutorial.
Some salient features of the structures:
| PDB ID | enzyme | family | chains | conserved residues | ||
|---|---|---|---|---|---|---|
| 1uyp | invertase, T. maritima |
GH32 | A-F | Asp17 | Asp138 | Glu190 |
| 1gyd | arabinanase A, C. japonicus |
GH43 | B | Asp38 | Asp158 | Glu221 |
| 1oyg | levansucrase, B. subtilis |
GH68 | A | Asp86 | Asp247 | Glu342 |
First, simplify the situation by deleting unwanted (for our purposes) portions of the structures such as extra chains and solvent, and then displaying just the alpha-carbon traces.
Command: delete #0:.b-fSince a set of structurally equivalent residues is known, an efficient way to superimpose the structures is by specifying atoms to use in a least-squares fit:
Command: delete solvent | ions
Command: chain @ca
Command: linewidth 2
Command: match #1:38.b,158.b,221.b@n@ca@c@o #0:17.a,138.a,190.a@n@ca@c@oIn fact, the chain specifications .a and .b could be omitted from these match commands because the residues would still be specified uniquely without them (other chains containing the same residue numbers in the same model have been deleted). In the general case, chain identifiers are needed to specify residues uniquely because a given residue number may occur in more than one chain.
Command: match #2:86.a,247.a,342.a@n@ca@c@o #0:17.a,138.a,190.a@n@ca@c@o
The peptide backbone atoms of the conserved residues were used to match the structures. Another possibility would have been to use all atoms of these residues, as the residue types are the same across the different structures (match requires equal numbers of atoms from the two models being matched). Model 0 (invertase) was used as the reference structure for both pairwise matches. The number of points used for fitting and the resulting RMSD values are reported in the status line (transiently) and the Reply Log.
Alternatively, matchmaker could have been used to superimpose the structures. Instead of user-specified atoms, it uses the protein sequences and secondary structure to generate alignments. Superpositions similar to those obtained above with match can be obtained with the following:
Command: matchmaker #0 #1Using matchmaker is usually much easier than using match because structurally equivalent atoms do not need to be specified. However, matchmaker:
Command: matchmaker #0 #2
Undisplay atoms and show the structures as ribbons:
Command: ~dispThe default ribbon style is flat. The edged and rounded styles are more three-dimensional.
Command: ribbon
Command: ribrepr roundedThe Chimera default ribbon scaling (secondary-structure-specific height and width) was used for the example images. New scalings and styles can be defined, named, and saved for later use with the Ribbon Style Editor (Tools... Depiction... Ribbon Style Editor).
The entire protein chains are shown. Although not done for the examples, ribbon can be undisplayed for parts of chains:
Command: ~ribbon #0:296-end
Select the conserved residues:
Command: select #0:17,138,190 #1:38,158,221 #2:86,247,342The atom specification should not contain spaces except (optionally) before the model symbols #. As explained above, chain specifications are not required; although :17.a could be used, :17 is sufficient.
Display the residues as ball-and-stick:
Command: disp selEven though all atoms of the residues are "displayed," by default the backbone atoms are hidden when ribbon is shown (this can be controlled with the command ribbackbone).
Command: repr bs sel
Command: ~sel
Color the three models three distinguishable colors.
Command: sel #0Repeat for models 1 and 2. For the example images, the colors named coral, medium purple, and turquoise were chosen for models 0, 1, and 2, respectively.
Actions... Color... all colors [pick a color]
The background can be any color, but white is often good for publication. Clear the selection and make the background white:
Command: ~selOther ways to change the background color are with the menu (Actions... Color), the Background preferences, or presets. Presets change several display settings at once.
Command: set bg_color white
Resize the Chimera window to the desired aspect ratio and find view(s) suitable for the images. A view or position refers to the locations and orientations of the structures, the overall scale, and the positions of the clipping planes.
For the example images, a position was found that showed the overall structures well, and when scaled up (without further rotation) showed the conserved side chains well. The zoomed-out position was named overall and the scaled-up position was named closeup:
Command: savepos overallSaved positions are included in saved sessions and can be regenerated:
Command: scale 4
Command: savepos closeup
Command: reset overallThese positions were used for the example images.
Command: reset closeup
The start command provides an alternative to choosing tools from the menu. For example, open the Shininess tool:
Command: start ShininessSet the shininess to 128 and the brightness to 5. Although not done for the examples, the positions, colors, and intensities of the two light sources in Chimera can be adjusted. Close the tool.
Open the Effects tool (Tools... Viewing Controls... Effects). Depth cueing is shading the scene from front to back, which looks like "fog" when the depth cueing color is light. The default depth cueing color is undefined, meaning that it will automatically match the background. The amount of depth-cueing shading that is visible can be adjusted by moving the clipping planes in the Side View (another tab in the same interface that includes Effects). Turn off depth cueing to remove the fog effect.
Also in Effects:
A session file can be saved with File... Save Session As (in this case, images.py was saved). It is generally prudent to save sessions for publication images, as this decreases the labor necessary if figures have to be redone. Often minor adjustments such as changing a color or displaying a different set of side chains will be required.
Later, the session can be restarted with File... Restore Session. Any positions that were saved before the session was saved can be regenerated with the command reset.
Choosing File... Save Image brings up the Save Image panel.
Supersampling refers to generating an initial image larger than that requested and then sampling it down to the final size. This smooths edges within the image. The default Supersample setting of 3x3, used for the examples, is generally sufficient for publication.
Maintain current aspect ratio means that if the size along one dimension (width or height) is changed, clicking the entry field for the other dimension will automatically update its value according to the current aspect ratio of the graphics window. The dimensions can be specified directly in pixels, or in units of length (when Use print units is on). If units of length, the output pixel dimensions are calculated using the specified Print resolution.
To make an image for a single-column width of 85 mm (3.346 in) with a resolution of at least 300 dpi, possible approaches include:
The example images were saved in TIFF format; other possibilities are PNG, JPEG, PS (PostScript), and EPS (Encapsulated PostScript).
Labeling, stereo, and color space are discussed in the Tips on Preparing Images. Images can also be created by raytracing with POV-Ray.