

 home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search

Comparisons among multiple proteins are essential for research on protein structure and function. Even when a single protein is the primary focus of a study, comparisons with a broader set of related proteins can provide valuable information on what aspects of sequence and structure are conserved. Conservation may reflect requirements for some shared aspect of function such as folding, catalysis, or binding.
Methods for linking sequence and structure data are also important for functional analyses. Many more sequences than structures are available, providing more complete information on conservation, but structural comparisons can provide information not available from sequence alone. Structural superpositions can reveal residue equivalences between proteins even when their sequences are too divergent to align with confidence. Further, different conformations of the same protein can yield insights into function.
To these ends, UCSF Chimera includes a suite of tools for:
These sequence/structure tools are not isolated but play important roles in other features, including superimposing structures shown with the structureViz Cytoscape plugin and determining residue equivalences for morphing. They also work seamlessly with other Chimera features: for example, a macromolecular complex could be modeled by fitting subunit structures into EM density, then sequence alignments could be used to evaluate whether the predicted interfaces are conserved. Although the sequence/structure tools were designed primarily for use with proteins, they also work with DNA and RNA.
- displaying sequence alignments and individual sequences, automatically linking them with structures
- calculating conservation of sequence (entropy, variability) and structure (RMSD) and showing the values on structures
- sequence similarity searching and retrieval using a BLAST web service hosted by the RBVI
- fetching UniProt annotations and mapping them onto sequence
- superimposing structures with or without pre-existing sequence alignments
- creating alignments using sequence and/or secondary structure information
- creating structure-based sequence alignments
- multiple sequence alignment using web services hosted by the RBVI
- homology modeling using a Modeller web service hosted by the RBVI
The sequence/structure tools are also used by various databases and web servers via the Chimera web data mechanism:
- The Structure-Function Linkage Database - Chimera Web data files are provided for viewing enzyme structures, active sites (Chimera sessions), and sequence alignments.
- ModBase - Choosing the "launch Chimera" option to show a modeled structure displays it along with the template structure and their pairwise sequence alignment.
- The ConSurf Server - Clicking the link to view results in Chimera displays the query structure and output multiple sequence alignment colored by the server's conservation measure, as shown below.
The sequence/structure tools are under continued development. These and related features are outlined below, with links to full documentation including descriptions of capabilities not mentioned in this page. See also:
Tools for integrated sequence-structure analysis with UCSF Chimera. Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE. BMC Bioinformatics. 2006 Jul 12;7:339.


Sequence Viewer
The Multalign Viewer tool displays individual sequences and multiple sequence alignments. Sequence alignments can be read from external files (several formats) or created by other tools in Chimera. Structures opened in Chimera are automatically associated with sufficiently similar sequences in the alignment. After association,
Various measures of sequence conservation and structural variation (RMSD) can be computed and shown above the sequences as histograms, and on the structures with color or worm radius. Secondary structure elements can be depicted as colored boxes or regions on the alignment. Regions can also be created by hand.
- mousing over a residue in the sequence shows its structure residue number
- selecting in the sequence selects residues in the structure(s) and vice versa
- structures can be superimposed using the sequence alignment

Coloring by Conservation
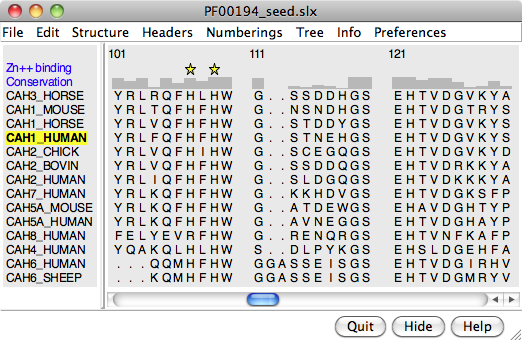
A structure can be colored to show attributes such as residue conservation. Opening a sequence alignment in Chimera shows it in Multalign Viewer and automatically associates sequences with structures as appropriate. Residues of alignment-associated structures are assigned conservation values; available measures include entropy, variability, and sum-of-pairs. The figure was created using the PFAM Carb_anhydrase seed alignment PF00194_seed.slx (see image) and includes 2D labels and a color key.
Superimposing Structures


There are several ways to superimpose structures in Chimera:
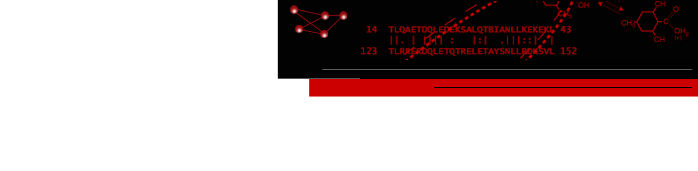
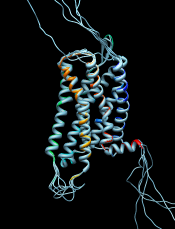
- MatchMaker performs a fit after automatically identifying which residues should be paired. Pairing uses both sequence and secondary structure, allowing similar structures to be superimposed even when their sequence similarity is low to undetectable. The figure shows five distantly related proteins (pairwise sequence identities <25%) from the SCOP WD40 superfamily before and after MatchMaker superposition with default parameters.
- Structures can be matched using a pre-existing sequence alignment.
- The exact atoms to pair can be specified with the match command. This works on any type of structure, while the preceding methods apply only to peptide and nucleotide chains.
- Structures can be superimposed manually by activating/deactivating them for motion and using the mouse.

Structure-Based Sequence Alignment
Given two or more superimposed structures, Match→Align creates a corresponding sequence alignment. The user specifies a distance cutoff for residues allowed to be in the same column of the output alignment. In proteins, the distances are measured between α-carbons. The method is independent of residue types and how the structures were superimposed.
The figure shows a superposition from MatchMaker of five proteins from the SCOP WD40 superfamily and a corresponding sequence alignment from Match→Align, automatically shown in Multalign Viewer. In the sequence alignment, light green and yellow boxes indicate strands and helices, while the headers RMSD and Conservation show spatial and sequence conservation, respectively.
Blast Protein
The Blast Protein tool performs a blast or psiblast search of pdb or nr for sequences similar to a query, using a web service hosted by the UCSF RBVI. The query can be:
The output is a list of hits, from which all or a user-chosen subset can be retrieved:
- a chain from a structure open in Chimera
- a sequence pasted as plain text
- a sequence from an alignment in Multalign Viewer
- as a pseudo-multiple sequence alignment (a consolidation of the pairwise alignments of individual hits to the query), automatically shown in Multalign Viewer
- as structures (for hits from pdb), automatically superimposed according to the pseudo-multiple alignment
Annotations from UniProt
PDB/UniProt Info retrieves sequence and structure annotations for Protein Data Bank (PDB) entries using a web service provided by the RCSB PDB. Sequences are displayed in Multalign Viewer, and feature annotations from UniProt are mapped onto the sequences as regions or colored boxes. In the region browser (figure at right):
- making a region Active selects any corresponding structure residues
- making a region Shown displays it in the sequence window
- the square color wells show (and allow changing) the region interior and border colors
UniProt annotations can also be fetched along with a sequence or mapped to a sequence already in Multalign Viewer regardless of whether the sequence is associated with a structure.


Multiple Sequence Alignment
Multiple sequence alignment of structure chains in Chimera or realignment of the sequences in an existing alignment can be performed using web services hosted by the UCSF RBVI. The following programs are provided:
The result is automatically shown in Multalign Viewer. (Sequences can also be added to an alignment one by one without a web service, but true multiple sequence alignment is often advantageous.)
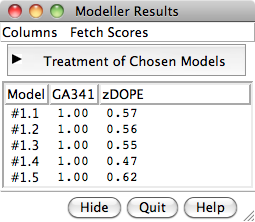
Chimera Interface to Modeller
Chimera provides a graphical interface to running the program Modeller, either locally or via a web service hosted by the UCSF RBVI. Two types of calculations are available:
Modeller is developed by the Sali Lab.
- Comparative (homology) modeling. Theoretical models of a protein target are generated using at least one known related template structure and a target-template sequence alignment. There are several ways to generate these inputs in Chimera. See also the Comparative Modeling tutorial.
- Building parts of a protein without using a template. Missing segments can be built de novo, or existing segments refined by generating additional possible conformations.

Showing ConSurf Results
The ConSurf Server provides results as Chimera web data; after browser configuration, a single click displays the color-coded query structure and multiple sequence alignment with phylogenetic tree and custom headers in a locally installed copy of Chimera (details).
Special thanks to Elana Erez and the Ben-Tal and Pupko groups at Tel Aviv University, and to Fabian Glaser at the Technion.
Laboratory Overview | Research | Outreach & Training | Available Resources | Visitors Center | Search



")


{kind=link}