Tom Goddard
March 15, 2011
Here is a sketch of the Chimera tutorial for the EMAN 2011 Workshop.
For full details view the screen capture video version of the tutorial.
Video tutorial| Data: |
We look at results from
Visualization of a missing link in retrovirus capsid assembly. | |
Steps:
|
Using Chimera version 1.5.2. |
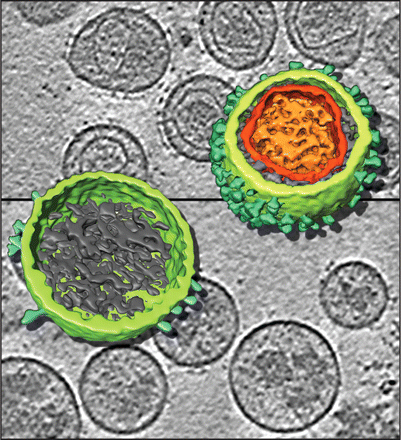
The map is an in vitro construction of empty virus capsids made from 60 copies of the Rous sarcoma virus capsid protein. Actual virus capsids have about 20 times more capsid proteins and are irregular polyhedra. Capsid protein is very similar in HIV.
Open map emd_1862.map using menu File / Open.... Or fetch map from web (slow) using menu File / Fetch by Id entering 1862 in EMDB entry field.
Set contour level in volume dialog so that enclosed volume of surface is 1.8 x 106 Å3, the expected volume for 60 copies of 25 kDalton capsid protein with ~1.2 Å3 per Dalton. Use volume dialog menu Tools / Measure Volume and Area.
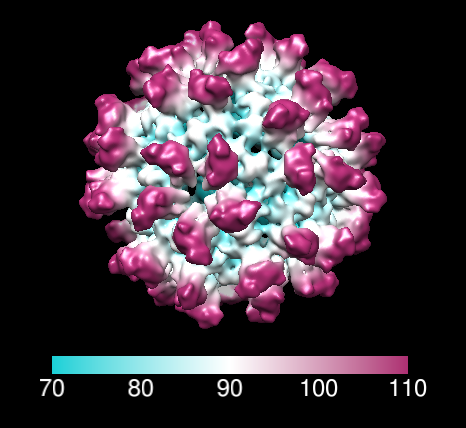
Color map radially using volume dialog menu Tools / Surface Color, press Color button. Can change colors and create color key using options shown by pressing Options button.
Fit crystal structure of N-terminal domain (NTD) of capsid protein into map.
Open 1em9.pdb using menu File / Open... or use menu File / Fetch by Id to fetch from web.
Hide map with eye button in volume dialog next to step size above histogram.
Use menu Presets / Interactive 1 (ribbons) to color chains. 1em9 is a dimer. Delete red chain B by selecting one residue (ctrl-click) then press up-arrow keyboard key to select whole chain, then menu Actions / Atoms and Bonds / delete. Use Presets / Interactive 1 (ribbons) again to color single chain as rainbow.
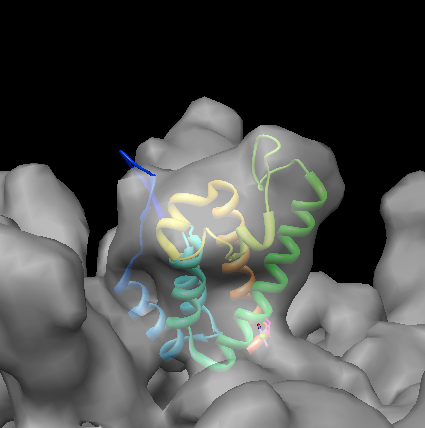
Move model to approximately correct location in map. Use Model Panel (Favorites menu), show map with checkbutton in S column, freeze map with checkbutton in A (active) column. Move 1em9 with mouse into pentamer lobe of density map. Use ctrl-key with middle mouse button to move 1em9 in or out of screen.
Use volume dialog menu Tools / Fit in Map, choose Fit 1em9.pdb and press Fit button to optimize fit (rigid rotation and translation). Average map value is average density map value over all atom positions.
Make map transparent by pressing Color button on volume dialog, adjust 4th slider (alpha), click Opacity switch to see 4th slider.
Show correlation coefficient using Fit in Map Options button, enable "Use simulated map, resolution 10", press Update button. Changing contour level of simulated map changes domain where correlation is calculated and can change correlation values 0.8 to 0.9.
Load tutorial session file emd_1862_1em9_1eoq_fit.py using menu File / Restore session... to get fit N-terminal and C-terminal domains, or fit C-terminal domain yourself, pdb 1eoq.
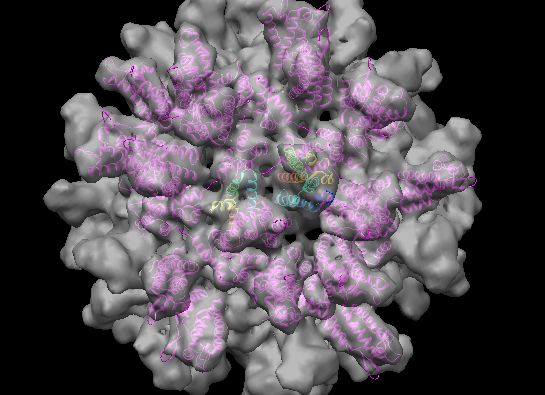
Create symmetric copies of fit crystal structure using sym command. Show command-line using menu Favorites / Command-line
sym #1 group i,222r coord #0 range 100
This command copies 1em9 fit model #1 (numbers listed in Model Panel) using icosahedral symmetry with standard reference orientation having 2-fold axes along x,y,z (222r) using the map (model #0) coordinate system, making copies only within 100 Å.
To copy both N-terminal and C-terminal domains, either repeat command for 1eoq (model #2) or combine domains into one model using:
~sym #1 combine #1,2 close #1,2 sym #3 group i,222r coord #0 range 100

Color map to show two neighboring capsid proteins. Select each protein ctrl-click then press up-arrow key to select entire molecule, then color with menu Actions / Color / orange. Color adjoining protein a different color.
Select both colored proteins using ctrl-click, shift-ctrl-click to add to selection, and up-arrow key. Use volume menu Tools / Color Zone, set radius to 5 and press Color to color map surface to match colors of selected molecules within 5 Å.
Hide all molecule using Model Panel (Favorites menu) click density map list entry and press Show only button in right column of Model Panel.
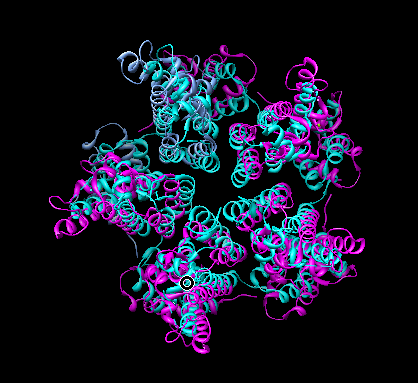
Compare HIV crystal structure pentamer 3p05 to fit Rous sarcoma virus pentamer.
Select Rous sarcoma virus pentamer using ctrl-click and shift-ctrl-click to select one residue in each of 5 molecules, then up-arrow key. Invert selection with menu Select / Invert (all models) and hide other molecules using menu Actions / Ribbon / hide and Actions / Atoms and Bonds / hide.
Open HIV pentamer 3p05.pdb with menu File / Open... or File / Fetch by Id....
Move and fit HIV pentamer into Rous sarcoma map. Select 3p05 in Model Panel list and press Activate only, move to approximate fit in map, use Fit in Map dialog to optimize fit, use Model Panel Activate All button to allow moving all models. Hide map.
HIV pentamer and Rous sarcoma pentamer have very similar folds (same alpha helical bundles) and alignments.