Docking Review 11/24/08
One purpose of this paper presentation is to explore
whether Chimera should be improved to better support docking programs
other than DOCK. The paper does discuss the
popularity of different docking programs, but
otherwise it is about the "science" of docking, a somewhat different topic.
To try to accommodate both, the presentation will be in two chunks:
- science-y content of this review paper and some newer docking work
- some limited data on docking in the Chimera user community
Review paper:
Sousa SF, Fernandes PA, Ramos MJ.
Protein-ligand docking: current status and future challenges.
Proteins. 2006 Oct 1;65(1):15-26.
Docking aims to predict the structure of a complex
from structures of the separated molecules.
Each part of docking represents a trade-off
between thoroughness/accuracy and computational demands:
- Searching:
sampling conformations, rotations, translations.
Originally the molecules were treated as rigid bodies.
Now most docking includes small molecule ligand flexibility,
and methods for handling (limited) receptor flexibility are emerging.
- Scoring:
evaluating which ligand conformation and
rotation/translation relative to the receptor
(which pose) is most likely,
and in virtual screening applications,
ranking different ligands by binding affinity.
Fast yet reasonably accurate scoring is the holy grail.
Search Algorithms (Flexible Ligand Docking)
- systematic methods
- conformational search
- "dihedral driver," sample all torsions exhaustively given some increment;
due to the potential combinatorial explosion, programs don't actually use
this approach
- fragmentation - either dock multiple fragments and then try to
connect them with bonds, or dock an anchor fragment and build
outwards in steps from that bound position
- database methods - just generate multiple reasonable conformations
of each small molecule ahead of time (stored in database), then dock them
as rigid bodies
- FLOG has this built in, but the same approach can be used with any
other program, you just have to create the database separately
- stochastic methods
- Monte Carlo (MC) - "randomly" tweak ligand conformation or
position relative to the receptor, accept the new pose with a probability
based on comparing the previous score to the current score. Good at
hopping energy barriers, and unlike MD,
does not require calculation of forces (the scoring
function does not have to be differentiable).
- genetic algorithm - start with a population of poses, where
descriptors of conformation and position relative to the receptor are the
"genes" and the score is the "fitness"; perform mutations, crossovers, etc.
of the fittest to create the next generation, repeat to convergence.
- tabu search - the ligand is repelled from previously explored
positions
- (I believe MORDOR does something like this)
- simulation methods - these are better classified as postprocessing
options rather than docking methods per se
- energy minimization
- ICM, DOCK, etc. have such an option
- molecular dynamics (MD) - the authors seem quite down
on this approach and say it is bad at hopping energy barriers;
compensatory strategies include simulating at high temperatures
and performing multiple runs for multiple starting poses.
Scoring Functions
- force field-based - containing terms similar to those
in force fields (AMBER, CHARMm, etc.) for VDW interactions, hydrogen bonding,
and/or Coulombic electrostatics.
Usually a distance-dependent dielectric is used to roughly mimic charge
screening by solvent water, which is not explicitly represented.
- GoldScore, AutoDock scoring, DOCK force field scoring, etc.
- empirical - based on regression analysis of a training set of
complex structures and their experimentally determined binding affinities,
typically containing terms that depend on functional group and type of
interaction (e.g. N-O hydrogen bond, O-O hydrogen bond,
salt bridge, aromatic ring stacking, etc.)
- LUDI score, ChemScore, AutoDock scoring, etc.
- knowledge-based - based on statistical analysis of training set
of complex structures to give distance-dependent pair potentials for atoms,
functional groups, or residues (a similar approach has been used to evaluate
the quality of homology models)
- PMF score, DrugScore, etc.
- consensus - simply combining the scores or rankings
from different scoring functions in various ways
(Protein) Receptor Flexibility
The older concepts of lock-and-key fit (rigid preorganization)
and induced fit (receptor adjusts as it binds ligand) have been replaced by
a picture where unbound structures form ensembles of conformations,
some of which resemble the bound conformation. Ligand binding stabilizes
those states and shifts the equilibrium among the different conformations.
Digressing from the 2006 review paper...
In case anyone is interested, here is a good
recent review on handling protein flexibility in the context of docking:
Andrusier N, Mashiach E, Nussinov R, Wolfson HJ.
Principles of flexible protein-protein docking.
Proteins. 2008 Nov 1;73(2):271-89.
(even though it is about protein-protein docking, most of it applies also
to the protein side of protein-small molecule docking; covers hinge-finding,
normal mode analysis, principal components analysis, sidechain placement
algorithms, implementations in docking programs, etc.)
Recent Docking Developments
An exciting recent development in docking is predicting the function
of an enzyme by identifying its natural substrates.
Functional annotation from structure is a huge and growing area of
investigation.
These successful predictions were enabled by identifying the
protein of interest as a member of a particular superfamily.
This allowed narrowing the possible substrates and types of reactions to
a reasonable search space. Different approaches were used
to accurately rank the docked molecules, however.
- Shoichet, Almo, Raushel groups (docking program DOCK 3.5.x):
instead of docking a database of metabolites, they
used knowledge that the amidohydrolase superfamily performed
hydrolysis reactions, and built a database of transition-state-like
structures based on hydrolysis-susceptible metabolites.
For example, esters and amides
were elaborated into tetrahedral forms, phosphoesters into
trigonal bipyramidal forms, etc. The idea is that since enzymes evolved
to catalyze reactions by binding the transition state better than the
substrate(s), docking transition-state-like molecules should provide
a better signal than docking substrates. The protein was kept rigid.
Structure-based activity prediction for an enzyme of unknown function.
Hermann JC, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, Raushel FM.
Nature. 2007 Aug 16;448(7155):775-9.
Predicting substrates by docking high-energy intermediates to enzyme structures.
Hermann JC, Ghanem E, Li Y, Raushel FM, Irwin JJ, Shoichet BK.
J Am Chem Soc. 2006 Dec 13;128(49):15882-91.
[journal club
presentation]
- Jacobson, Almo, Gerlt, Babbitt groups (docking program Glide): these
successes were obtained with comparative models rather than experimental
structures. Identifying the enzymes as belonging to a certain subgroup
of the enolase superfamily allowed narrowing the set of possible
substrates, and accuracy in ranking was improved by
refining and rescoring the docked complexes with a more complicated
physics-based scoring function and allowing receptor sidechains to move.
Discovery of a dipeptide epimerase enzymatic function guided by homology modeling and virtual screening.
Kalyanaraman C, Imker HJ, Fedorov AA, Fedorov EV, Glasner ME, Babbitt PC, Almo SC, Gerlt JA, Jacobson MP.
Structure. 2008 Nov;16(11):1668-77.
Prediction and assignment of function for a divergent N-succinyl amino acid racemase.
Song L, Kalyanaraman C, Fedorov AA, Fedorov EV, Glasner ME, Brown S, Imker HJ, Babbitt PC, Almo SC, Jacobson MP, Gerlt JA.
Nat Chem Biol. 2007 Aug;3(8):486-91.
Popularity of Different Methods
It is difficult to compare protein-ligand docking programs. There is even
a paper with that title. Nevertheless, many have tried.
A couple of recent examples:
Comparative evaluation of eight docking tools for docking and virtual screening accuracy.
Kellenberger E, Rodrigo J, Muller P, Rognan D.
Proteins. 2004 Nov 1;57(2):225-42.
(compared Dock4, FlexX, Fred, Glide, Gold, Slide, Surflex, QXP
and found that Glide, Gold, and Surflex were the most successful)
A critical assessment of docking programs and scoring functions.
Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS.
J Med Chem. 2006 Oct 5;49(20):5912-31.
(compared 10 docking programs and 37 scoring functions;
results varied widely for different receptors, but in general
Dock4 performance was poor)
Back to the 2006 review paper...
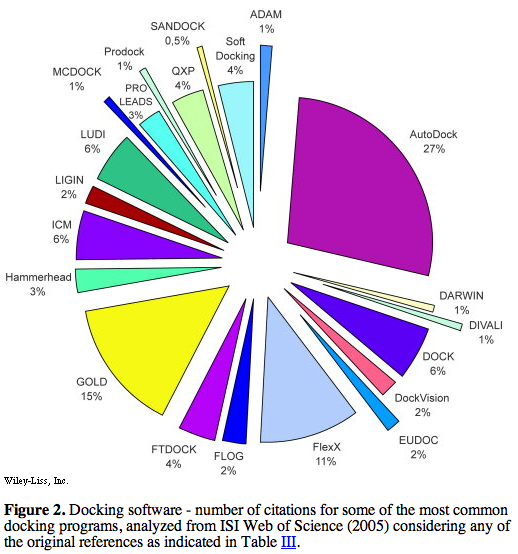
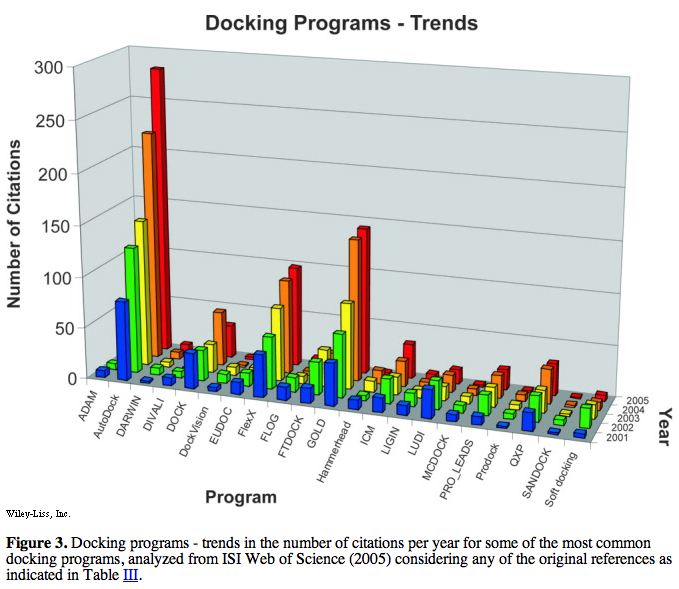
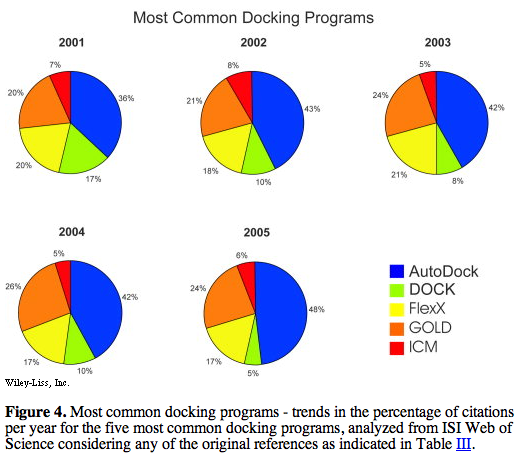
prevalence of use according to literature citations was presented in figures:
Of the top five (above), only AutoDock and DOCK are free to academics.
Chimera Users and Docking
I had saved an old
user
e-mail exchange on this topic.
I looked through my
Chimera
citations 2008 file for notes about use of docking programs.
The notes were intended to describe how Chimera was used, not necessarily
any other programs, so these counts are a lower bound.
Also, even though each paper both cited Chimera and described use of
a docking program, Chimera wasn't necessarily used to view the docking results.
- 4 uses Autodock
- 3 uses Gold (our buddy JD Marechal is an author on one of those papers)
- 1 use FlexX
- 1 use Patchdock (via the
Patchdock server )
- 1 use Glide
(probably disregard since in this paper Viewdock was used to view DOCK results,
Maestro to view Glide results)
- 1 use Haddock
(probably disregard since this protein-protein docking method was just
used to model one particular complex, not to screen multiple structures)
For comparison, there were 3 uses of DOCK (2 of those probably also
used Chimera ViewDock) and 1 MORDOR paper (also mentioned ViewDock)
out of 401 papers total listed.
In addition, I recently corresponded with Sebastian Kruggel, who uses
both FlexX and Autodock, and Eric recently corresponded with Sergio Marques,
who uses Gold. Neither is an author on any of the citations counted above.
More Tidbits
Autodock is free (GNU GPL), but
registration is required. There is an associated graphical interface,
AutoDock
Tools (ADT). AutoDock can allow protein sidechain flexibility.
It uses
PDBQT format,
basically a decorated PDB format similar to PDBPQR.
There wasn't a handy example file, just that cut-paste-unfriendly display,
but either Chimera reads it and just complains about bad records or
it is relatively easy for users to chop off the extra stuff
and read it as regular PDB. There are
tutorials
with sample input and output files supplied as tar.gz.
UCSF DOCK is free for academics, but you have to fill out a
licensing form.
DOCK 6 can allow receptor flexibility.
Gold
is from the Cambridge Crystallographic Data Centre. It can allow
partial protein flexibility (backbone and sidechain flexibility
of up to 10 residues).
FlexX
is from BioSolveIT and can be interfaced with
MOE
and SYBYL (Tripos).