- seven enzymes from the amidohydrolase superfamily, thus constraining the
reaction space to hydrolysis
(Scheme 1):
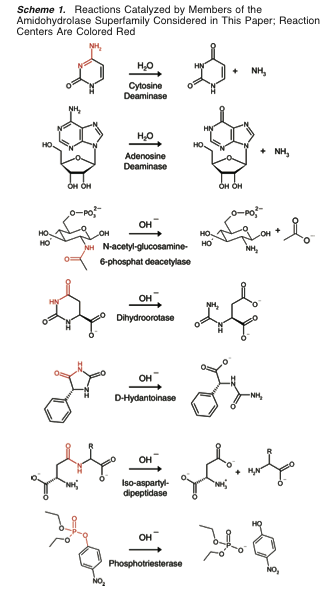
Predicting Substrates by Docking High-Energy Intermediates to Enzyme Structures. Hermann JC, Ghanem E, Li Y, Raushel FM, Irwin JJ, Shoichet BK. J Am Chem Soc 2006 Dec 13;128(49):15882-15891.Rationale:
Enzymes have evolved to catalyze reactions, not merely to bind substrates. Catalysis can be thought of as effective binding of transition states, where this favorable binding energy drives the reaction forward. Thus, to predict what reaction is catalyzed by an enzyme, it may be smarter to dock structures that resemble transition states than to dock substrates.Background:
Receptor structures:Docking to predict function has previously used databases of metabolites representing possible substrates and products. Docking transition states is probably not a new idea (considering that catalytic antibodies are generated with transition state analogs), but there are confounding issues.
- Generally, the geometries and charge distributions of transition states in enzymes are not known. Quantum mechanical calculations could be done, but the enzymes themselves dramatically affect the details of the transition state; without preknowledge of each enzyme's mechanism (which is what one is trying to predict anyway), only rough approximations can be used to construct the dockable database.
- Without some idea of the type of reaction catalyzed, the problem may not be sufficiently constrained, since many possible transition states could be generated from a given substrate.
The issues have not been solved here, but reasonable approaches have been used. A limited reaction space is involved, and the authors have been careful to use terms like high-energy intermediate and transition state analog as opposed to just transition state.
"Ligand" databases:
- seven enzymes from the amidohydrolase superfamily, thus constraining the reaction space to hydrolysis (Scheme 1):
Docking setup and parameters:
- "ground state"
- start with KEGG metabolite database
- supplement with dipeptides, hydantoins, phosphate esters
- retain only molecules with <48 atoms (not counting hydrogens)
- retain only molecules with hydrolysis-susceptible substructures → 3770 molecules
- "high energy"
- start with ground state database
- elaborate each metabolite at hydrolysis-susceptible substructure(s) (Scheme 2) → 21,000 high-energy compounds
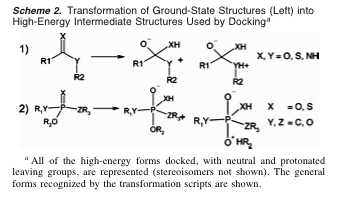
- construct flexibases (multiple conformations of each compound)
- generate different protonation states of groups with estimated pKa values in the range 5–9
- calculate partial changes and desolvation energies with semiempirical quantum mechanics
Retrospective docking (validation):
- AMBER united-atom parameters used for receptors
- +2 charges on metal ions partly redistributed to ligating residues
- attacking hydroxide or water included for docking to ground-state compounds, removed for docking to intermediates
- DOCK 3.5.54 with high sampling: up to a million poses per compound, multiple conformations scored per pose
- scoring: DelPhi electrostatic interaction + AMBER-style van der Waals interaction – prorated desolvation penalty
- rigidly minimize the best-scoring pose of each compound
- poses assessed for "catalytic productivity," whether the reactive substructure is within 3.5 Å of the attacking oxygen (ground-state compounds) or the former attacking oxygen is ligated by a metal ion (high-energy compounds)
- keep only the best-scoring intermediate elaborated from a given substrate
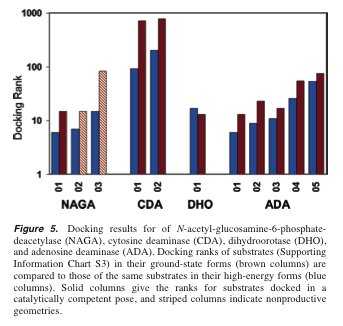
Prospective docking (prediction):For 5/7 amidohydrolase structures, docking the high-energy states outperformed docking the ground states. For dihydroorotase and adenosine deaminase (Fig. 5, below) both approaches worked well, with negligible differences. Enhancement seemed to be more pronounced for apo structures (those crystallized without ligand).
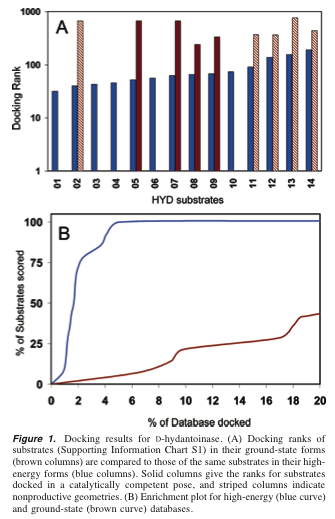
Results for D-hydantoinase, 14 known substrates in database (Fig 1):
high-energy vs. ground state docking
solid bars for catalytically competent poses
lower bars better
Does no bar mean not in top 1000?
Does the enrichment plot include both types of poses?
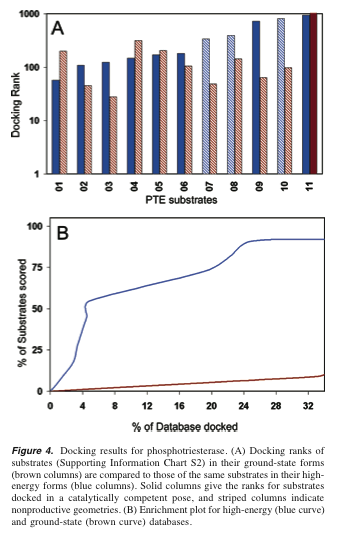
(brown line suggests it does, seems to end higher than 4/14)[ Not shown here: Stereoviews of 5-hydroxyethyl-hydantoin docked (Fig 2). ] [ Not shown here: Results for iso-aspartyl-dipeptidase, 37 substrates (Fig 3). ] Results for phosphotriesterase, 11 substrates (Fig 4): What's up with the curves?? Smoothing gone overboard? Fig 4B must not include both types of poses. Otherwise, both curves would be at 100% for 25% of the database docked and the brown curve would be above the blue curve. Upon careful re-examination, the descriptions of Figs. 3 and 4B hint at this rather obliquely.
Results for the other four, with relatively few substrates (Fig 5): [ Not shown here: Stereoviews of cytosine docked (Fig 6). ]
Five mutant phosphotriesterases (mutations at 1–3 active-site residues) were modeled by simply replacing the affected side chains. Stereoisomers of four potential new substrates (Chart 1) were docked into the wild-type and mutant receptors. The compounds were synthesized and tested "in real life." Of 24 possible predictions of enantioselectivity (6 receptors x 4 substrates), no predictions were made for 3 combinations because no catalytically competent pose was obtained. 19/21 predictions were qualitatively verified, two were wrong.
As far as I could tell, this part of the paper did not involve docking of high-energy intermediates.