

Several molecular display styles (representations) are available in Chimera. Atoms/bonds can be shown as:
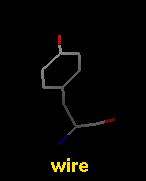
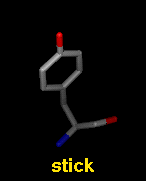
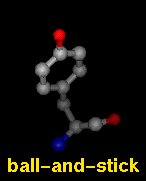
A model can only have one wire linewidth, but individual atoms and bonds can be shown in different representations. Atom and bond displays can be combined with ribbons and surfaces. See also: presets, New Molecules preferences
The command represent sets atom/bond styles, bondrepr sets bond styles, and linewidth changes wire linewidth. Atom/bond styles and wire linewidth can also be changed using the Actions menu, the molecule model attributes panel, and the Selection Inspector. Ball display radius is the product of the atomic VDW radius and the ball scale attribute of a molecule model (default 0.25). Stick radius is the product of the individual bond radius (default 0.2 for all bonds) and the stick scale attribute of a molecule model (default 1.0). Ways to change attribute values include the command setattr and the Selection Inspector. Different linewidth, ball-scale, and stick-scale values can also be specified in the New Molecules preferences.
Pseudobonds are drawn to show connections other than covalent bonds. For example, they can represent clashes, hydrogen bonds, or distance measurements. Pseudobond styles can be changed using the pseudobond attributes panel and the Selection Inspector.
In addition:
Protein and nucleic acid chains can be shown with ribbons. Protein helix and strand assignments are taken from the input structure file or generated with ksdssp. For nucleic acids, the ribbon simply follows the backbone. See also: PipesAndPlanks, Nucleotides
|
|
|
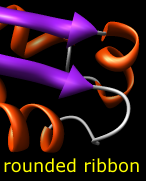
Built-in ribbon styles include:
The ribbon path is interpolated and may deviate from the exact positions of the backbone atoms (more...). The method of path calculation can be set with ribspline or in the molecule section of the molecule model attributes panel or Selection Inspector.
The insides of ribbons in protein helices can be colored separately using the Color Actions dialog, the molecule model attributes panel, or the command setattr.
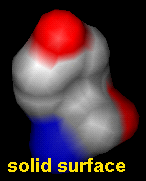
Chimera shows solvent-excluded molecular surfaces, composed of probe contact, toroidal, and reentrant surface. These differ from solvent-accessible surfaces, which are traced out by the probe center.
 |
 |
 |
Molecular surface styles are:
The atoms in a molecular model are automatically partitioned into categories for surfacing: ligand, ions, solvent, or main. By default, protein and nucleic acid chains are classified as main, and if multiple chains are in contact, they will be enclosed in a single surface. However, if separate surfaces for the separate chains are preferred, either of the following can be used:
Analytical solvent-excluded and solvent-accessible surface areas per atom and residue are assigned as attributes named areaSES and areaSAS, respectively. These may include contributions from more than one component. For example, one side of an atom could form part of an interior cavity while the other side could form part of the exterior surface. Calculating relative exposure (a normalized surface area) may be helpful for classifying amino acid residues as buried or exposed.
The command surface displays molecular surface, surfrepr sets which style is shown, and surftransparency adjusts surface transparency. Molecular surface display, style, and transparency can also be controlled with the Actions menu, the molecular surface attributes panel, the Selection Inspector, and the command setattr. Except for the Actions menu, these also allow changes in probe radius, vertex density, mesh line width, and dot size. Parameters for subsequently generated molecular surfaces can be set in the New Surfaces preferences.
In Chimera, molecular surfaces are created with embedded software from the MSMS package, described in:
Reduced surface: an efficient way to compute molecular surfaces. Sanner MF, Olson AJ, Spehner JC. Biopolymers. 1996 Mar;38(3):305-20.MSMS surface calculations may fail numerically, especially on large structures; see surface calculation failures and workarounds.
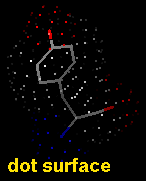
Dot molecular surfaces in MS/DMS format can also be displayed in Chimera.
A van der Waals (VDW) surface differs from a molecular surface in that fine crevices are not smoothed. A VDW dot surface can be displayed with the command vdw and its dot density adjusted with vdwdensity. The VDW dot size and dot density can also be adjusted in the the molecule model attributes panel. Note that the sphere representation also shows the VDW surface.
Chimera also displays other surfaces that are not necessarily molecular.