Visualization and Analysis Challenges of Large Biomolecular Complexes
Tom Ferrin
Resource for Biocomputing, Visualization and Informatics
University of California, San Francisco
This slide deck available at:
www.rbvi.ucsf.edu/home/tef/talks/MolecularMachines.html
Background
Since 2009, there has been a rapid increase in number of available models of complex systems. These systems are often composed of dozens or even hundreds of proteins, frequently built using multiple experimental methods at varing resolutions and including flexible regions with poor structural detail, thus posing several challenges to the interactive visualization and analysis of their 3-D structures.
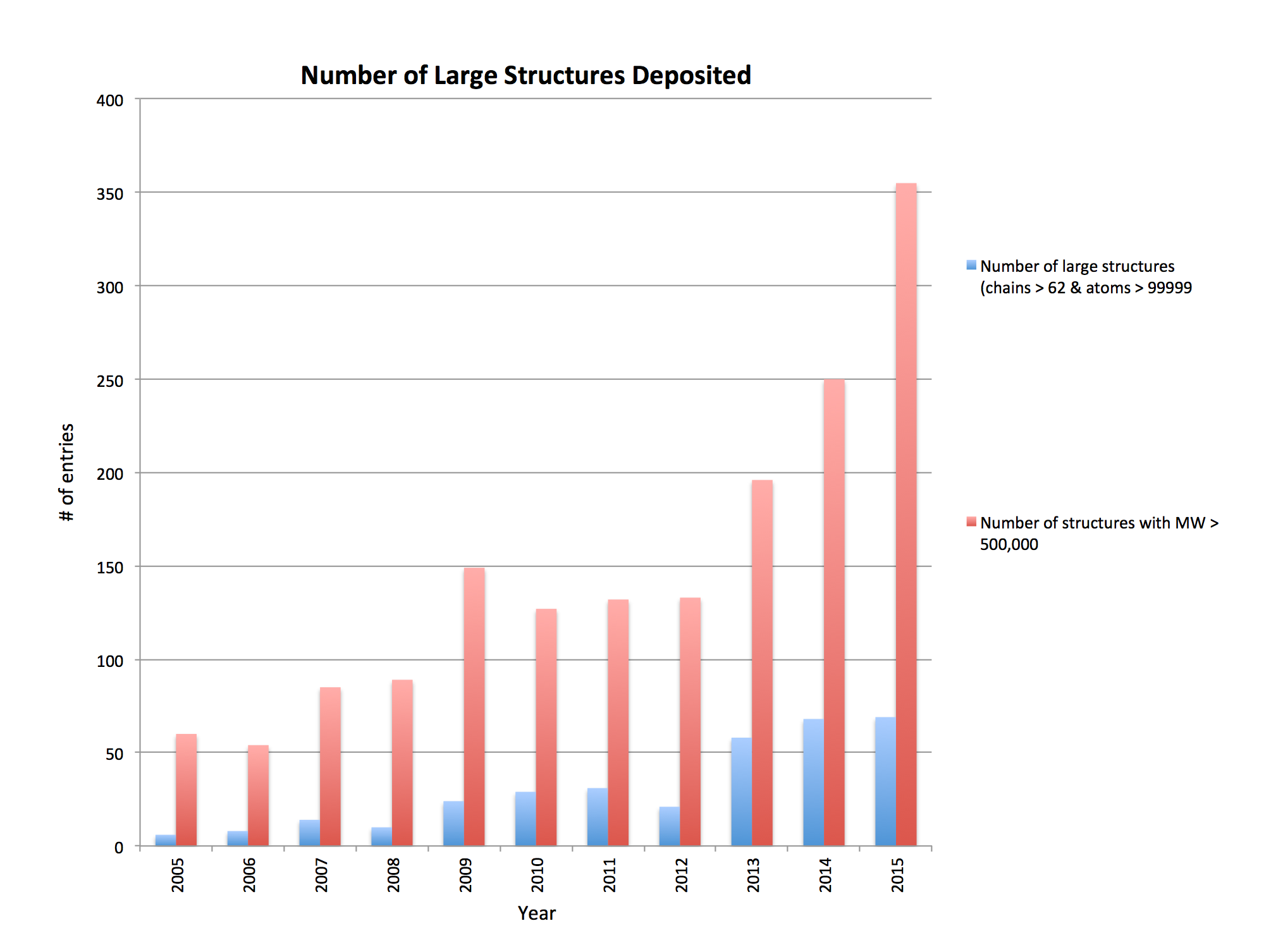
Source: Jasmine Young, RCSB
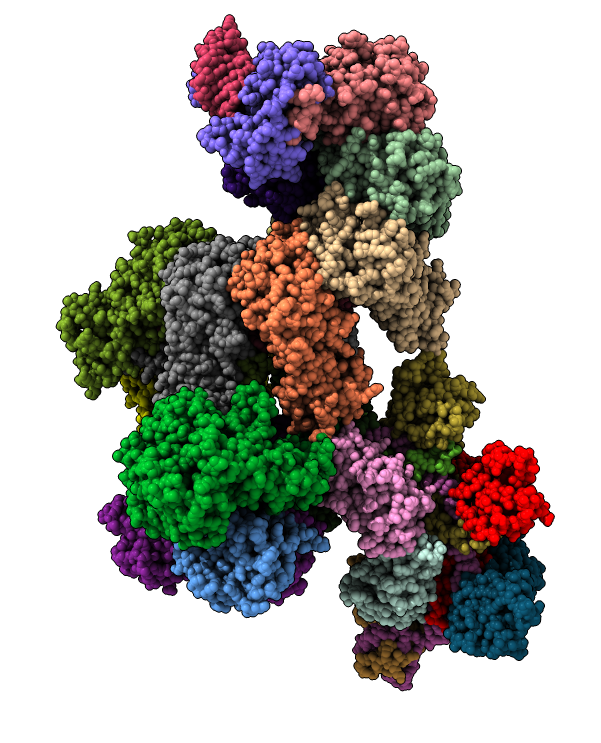
Example 1
CRISPR: 5cd4. Two copies of the complex contained in the crystal asymmetric unit and defined in mmCIF assembly table. Each complex consists of 6 copies of CasC, 2 copies of CasB, 1 copy each of CasA, CasD, CasE (latter have identical sequences). 53,308 atoms.
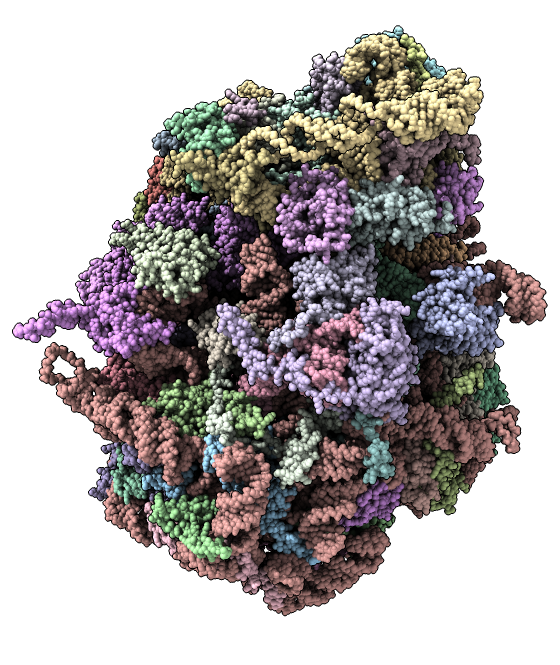
Example 2
Ribosome: 4v6w. 80 protein chains, 6 nucleic acid chains, 230,721 atoms.
Limitation: Because there are more than 62 chains and 100,000 atoms must use mmCIF data format.
Problem: Functional components not defined in mmCIF, so no way to visually explore things such as "large subunit" or the "peptide synthesis site" except by chain ID.
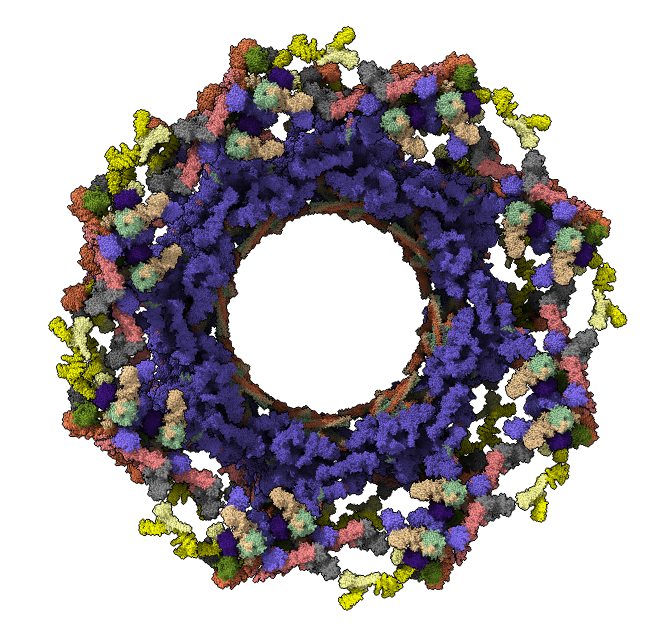
Example 3
Nuclear pore complex: 736 copies of 20 proteins with 11 x-ray structure complexes (5HAX 5HAY 5HAZ 5HB0 5HB1 5HB2 5HB3 5HB4 5HB5 5HB6 5HB7 5HB8) fit into a cryoEM map (EMDB 3103), 8-fold symmetry, ~4,000,000 atoms.
Question: How would you find all contacts between 2 of the 8 "spoke" complexes, or between all channel and adaptor proteins?
Challenges and Opportunities
- Diverse data types from multiple experimental methods calling for versatile data repositories
- Multiple levels of data organization (hierarchies, groups, functional entities...) requiring richer data relationships than currently defined and new graphical user interfaces for navigating these relationships
- Very large multi-million atom models necessitating memory-efficient data structures and high performance graphics
ChimeraX Demo
(Length: ~15 minutes)
Part A: Nuclear pore electron microscopy
- Nuclear pore (human) composed of 30 different proteins, ~1000-copies, 8-fold symmetry, 23Å resolution
- Special GPU "shader" program provides ambient occlusion lighting with shadows emphasing crevices for better depth perception
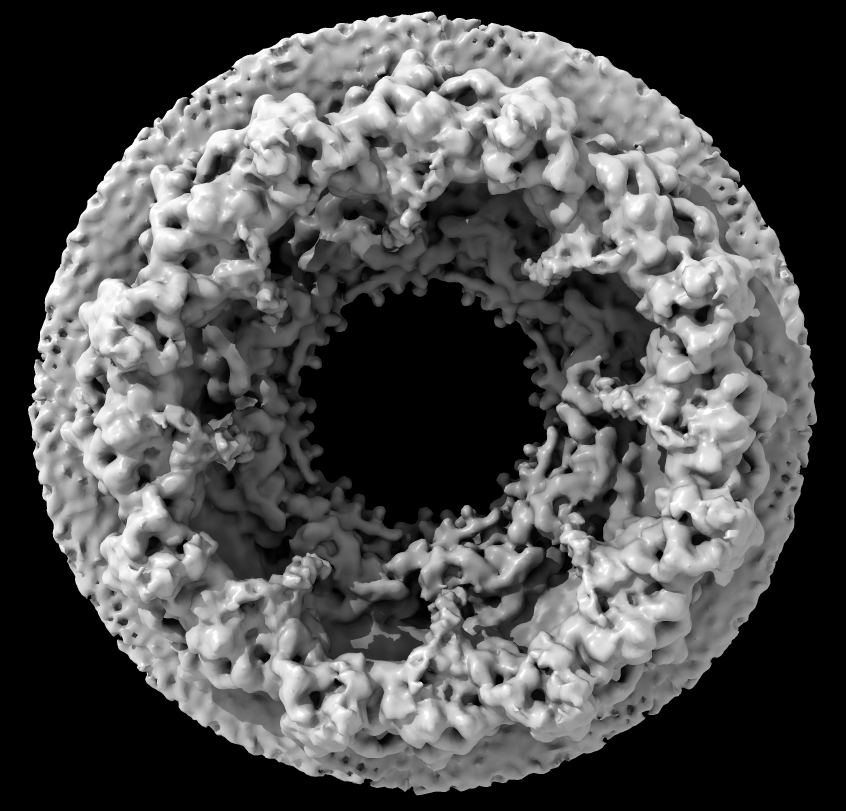
Source: Martin Beck lab, EMBL Heidelberg
Part B: Nuclear pore atomic model
- 736 copies of 20 proteins; 4 million atoms composing about half the mass of the nuclear pore, fit to EM density map
- Hierarchical organization: inner and outer rings, spokes, and Y-complex. Asymmetric unit has 92 proteins
- Copies of Y-complex form scaffold for outer rings
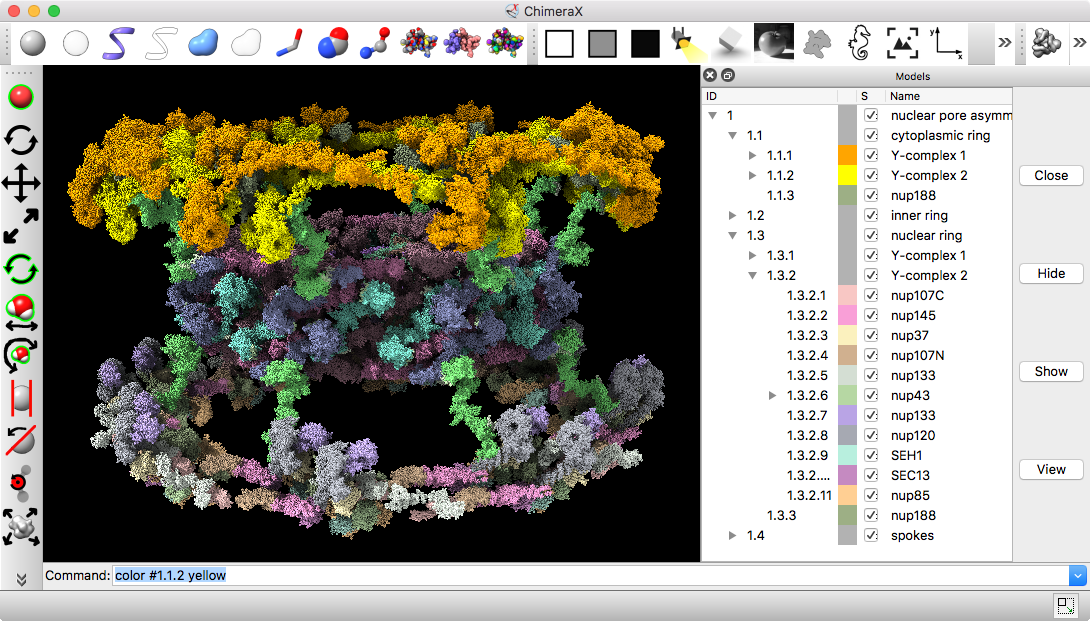
Source: Andre Hoelz lab, Cal Tech
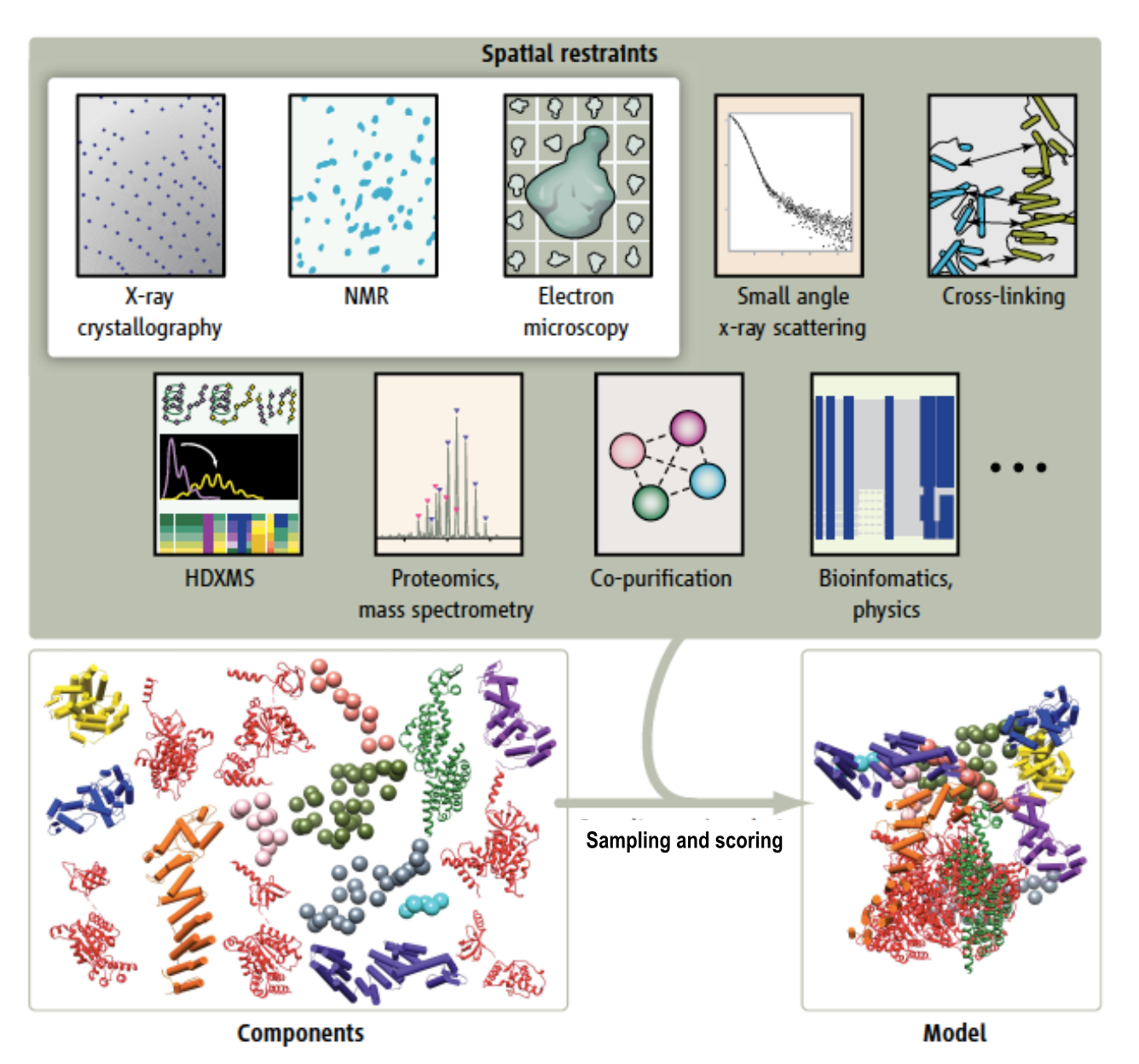
Part C: Integrative hybrid model of Y-complex
- 7 protein Y-complex (also called nup84) forms scaffold for nuclear pore outer rings
- Model built from many data sources:
- Two protein X-ray models
- Six comparative models, 16 template structures and sequence alignments
- 300 chemical crosslinks resolved by mass-spectroscopy, 2 different linkers
- 2-D electron microscopy constraints model projection
- Disordered regions modeled with beads representing 10 residue segements
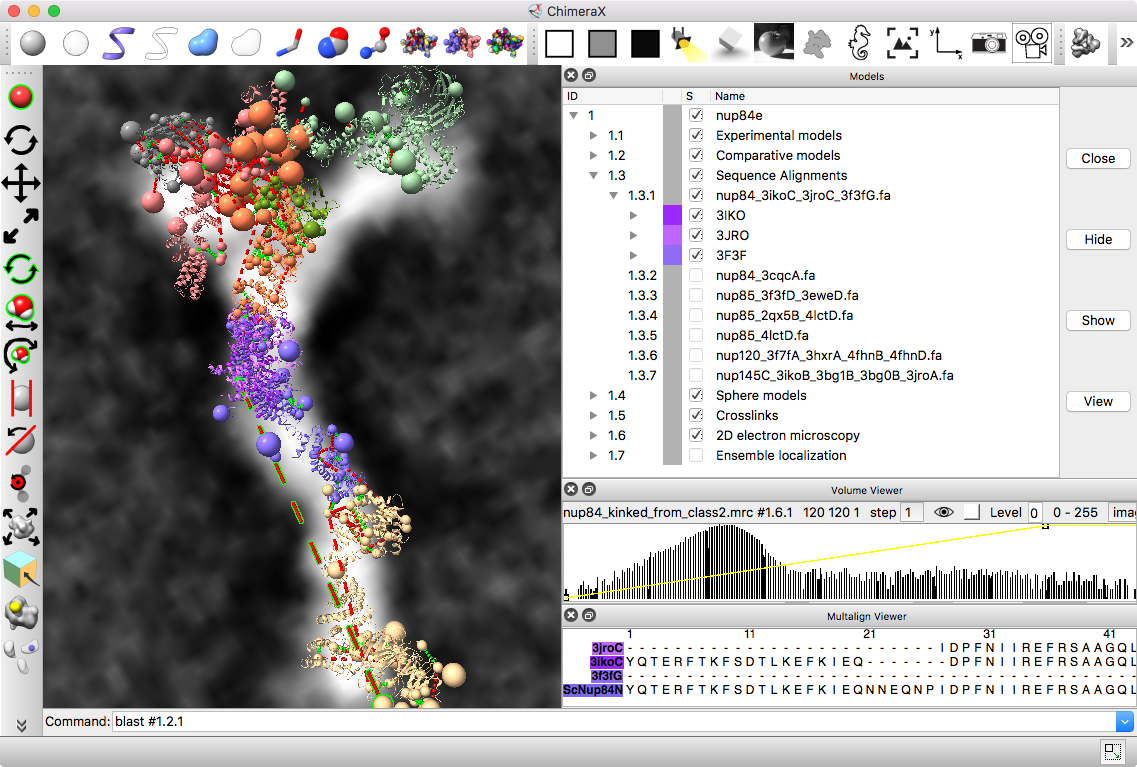
Source: Andrej Sali lab, UCSF
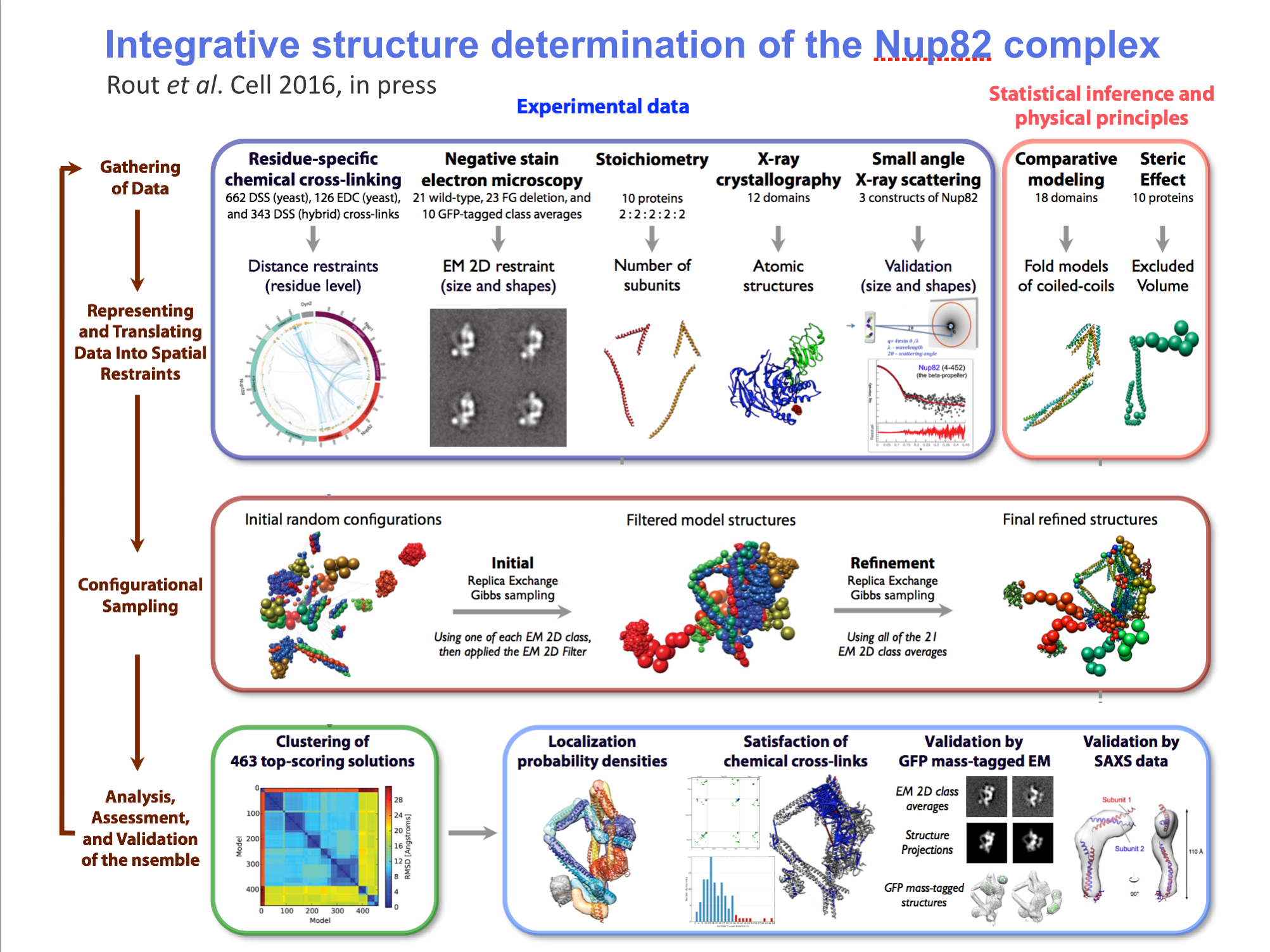
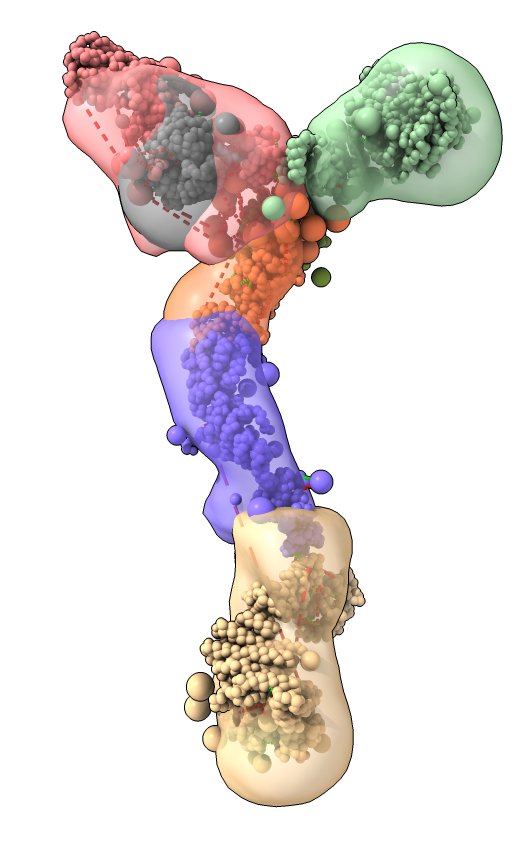
Model ensemble from sampling and scoring restraints
- Integrated Modeling Platform (IMP) software computes ensembles of thousands of models scored based on restraints
- Two clusters for two conformations each contain 1000 models
- Playing ensemble like one would a dynamics trajectory shows variation within ensemble
- Localization density maps for each protein in ensemble also shows variability
Integrative/Hybrid Models
- How should integrative models be represented?
- How should the data and models be validated?
- How should the data and models be archived?
- What information should accompany the publication of these models?

Integrative Models Task Force Workshop, EBI, October 2014
The Bottom Line
- Traditional PDB format structure files are obsolete and completely unworkable for large protein complexes, yet the majority of existing computational chemistry software still only understands this data format.
- Even the current mmCIF standard is inadequate for describing the many structural and functional data relationships present in important molecular machines such as the ribosome and NPC.
- Complementary to emerging mmCIF data extensions, richer graphical user interfaces are needed to navigate the multiple data relationships present in these machines.
- Innovation in interactive graphics rendering techniques, software data structures, and new user interfaces are necessary to facially manipulate large macro-molecular complexes.
