Journal Club 08/25/08
Levy ED, Boeri Erba E, Robinson CV, Teichmann SA.
Assembly reflects evolution of protein complexes.
Nature. 2008 Jun 26;453(7199):1262-5.
[PDF]
[suppl. PDF]
Summary:
Based on a large dataset of soluble (nonmembrane) homomeric complexes,
quaternary structure is well conserved, and changes that do occur
in evolution are biophysically logical. The order of assembly of multimers
may often parallel the path of evolution because in both cases,
larger interfaces tend to be more stable.
Background:
 |
The 3D Complex
database was described in a
2006 publication
by mostly the same authors.
It is hierarchical classification of complexes, meant to serve an analogous
role as the SCOP and CATH classifications of domains.
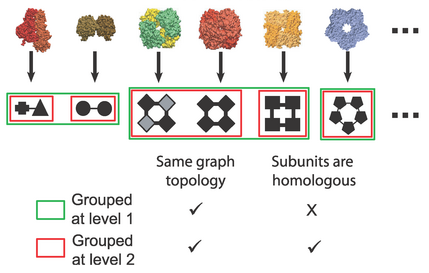
It includes graph depictions where a subunit is a node and a direct
interaction between subunits is an edge.
(figure at PLoS)
Levels in the hierarchy:
- quaternary structure topology (QST) - same number and arrangement of
nodes and edges; for example, all dimers would be in the same QST
- quaternary structure family (QSF) - each member's nodes are
homologous
to the corresponding nodes of the others, i.e. they share a
SCOP domain architecture
(have domains in the same SCOP superfamilies, in the same N→C order)
- quaternary structure (QS) - each member built from same number of gene
products
- further subdivisions redundant at different % ID
The edges are labeled with interface size (number of residues in contact,
averaged between the two chains).
As the graphs do not convey all symmetry information,
they are supplemented with Schönflies notation (more on this below);
however, symmetry is generally conserved within a QS.
Suspicious asymmetries in graphs led to the identification of
some probable errors in biological unit annotations from the
PDB and
Protein Quaternary Structure (PQS) server.
The PiQSi resource
(combination of "PQS" and "wiki" → pronounced "pixie")
containing ~10,000 biological unit annotations by Levy and a wiki-type
interface for additional annotation by the community was described in a
2007
publication. General annotation process:
- check primary citation for quaternary structure information
- check primary citations of any proteins 98-100% identical
- if >2 subunits, check if interface geometry is conserved with homologs
by superposition and visual inspection (relatively time-consuming but
provided conclusive evidence for many annotations)
- if still uncertain, further consult SwissProt, EcoCyc, PISA prediction
Of the ~10,000 manual annotations, ~15% disagreed with the PDB
biological unit annotation, comparable to the 16% error rate estimated
for PQS in 2003 by another group. Many errors show up as a lack of
symmetry, fairly easy to spot in the graph depiction.
PiQSi is now integrated with 3D Complex. (example:
1jpd)
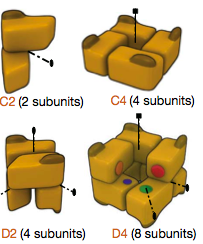
Terminology:
- open symmetry refers to proteins
that polymerize indefinitely (actin, tubulin)
- closed symmetry refers to complexes
with a defined number of subunits
- Cn
(Schönflies notation for crystallographic point groups)
refers to a cyclic complex with n subunits; n-fold rotation axis
of symmetry
- Dn refers to a dihedral complex with 2n subunits;
n-fold rotation axis plus a twofold axis perpendicular to that axis
Dataset:
5375 homomeric structures nonredundant at 80% identity
were taken from the manually curated set mentioned in the PiQSi paper.
This dataset is "~tenfold greater than any studied previously"
and relevant because most proteins with known QS are homomeric.
The dataset contains very few open complexes (3%, deemed harder to
crystallize) and complexes with cubic symmetry (1%);
membrane proteins are underrepresented.
2750 (51%) are monomeric, an important part of the dataset.
Observations and Discussion:
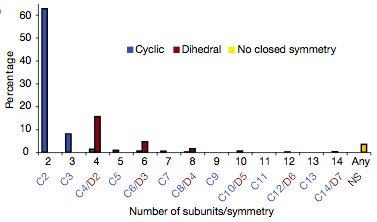
Consistent with earlier surveys of the PDB and SwissProt,
monomers are more common than dimers, dimers are more common than larger
complexes, and even numbers of subunits are more common than odd.
Dihedral complexes are much more common than cyclic complexes with
the same numbers of subunits but similar in prevalence to cyclic
complexes with half as many subunits.
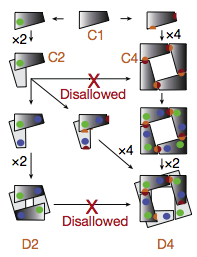
Presumably the original state was monomeric and multimers evolved in stages.
It is more likely for a single interaction patch to evolve at a time
than two separate patches. If it were necessary for 5 residues on A
to interact with 5 residues on B to form an AB complex, a face-to-face
or back-to-back interaction only requires evolution of a single suitable
5-residue patch, whereas a face-to-back interaction requires evolution of
two 5-residue patches. Dimerization requires a single patch, while
cyclization uses face-to-back interactions. Odd numbers of subunits in
a symmetric closed complex can only arise through cyclization.
Thus dimerization:
- monomer → dimer (C2)
- C2 → D2
- Cn → Dn (n>2)
is much more likely than cyclization:
- monomer → Cn (n>2)
- dimer (C2) → Dn (n>2)
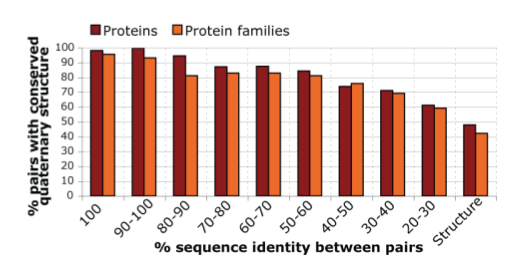
Quaternary structure (including symmetry) is highly conserved:
For each sequence identity range, a set of homologous structure pairs
was taken from the 3D Complex database.
Red bars show what fraction of pairs have the same quaternary structure.
Orange bars are analogous except considering only one pair per family.
It was not clear whether “family” meant QSF or something else.
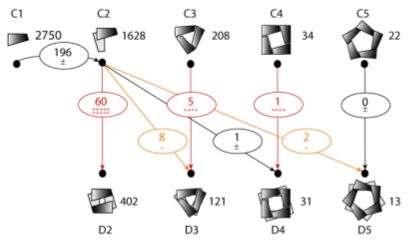
Comparing homologs with different quaternary structures shows that
dimerizations are more likely and cyclizations less likely
than in a random model of transitions.
Their explanation of the random model confused me.
“TQS
is the quaternary structure size (number of proteins)”
might be interpreted as the number of subunits, number of proteins
with that quaternary structure, or a product of the two,
and the superscript looks like an exponent at first. My best guess:
A first quaternary structure type is picked with a probability equal to
the number of proteins with that type divided by the total number of proteins.
A second quaternary structure is chosen in the same way, but with the first
type set aside so it cannot be selected again. After 100 such trials,
the average and standard deviation of the number of links
between each pair of types was calculated.
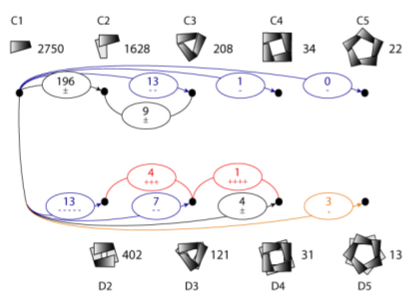
What about the order of transitions? Different paths can generate
the same higher-order complex. For example, a D3 complex could arise
by cylization of three dimers or dimerization of two trimeric rings.
The authors postulate the two paths are about equally likely because each
includes one dimerization and one cyclization. Energetic considerations
suggest that either way, the largest interface should form first,
allowing examples of the two paths to be distinguished.
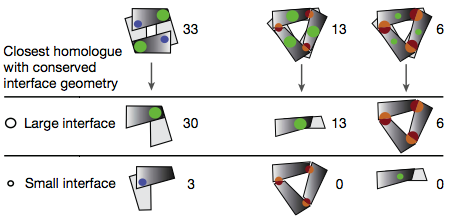
In fact, comparing related dimer-tetramer, dimer-hexamer,
and trimer-hexamer pairs shows greater conservation of the larger interface:
The energetic argument is that the loss of entropy is about the same in each
successive stage of multimerization. However, earlier steps form
fewer interfaces, so each interface must be bigger to compensate for
the total entropic loss.
Predicting evolutionary intermediates:
- group interfaces related by symmetry
- remove set with smallest average size (number of contacting residues)
- see what is still connected
- if any interfaces remain, go to #2
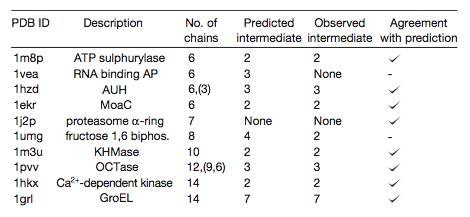
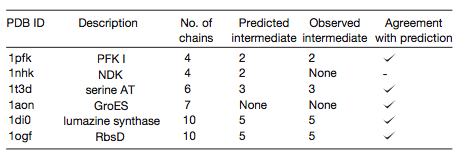
Ten complexes with predicted evolutionary intermediates were disassembled
by varying the ionic strength or adding denaturants, and
subcomplexes were detected by electrospray mass spectrometry.
In 8/10 cases, the disassembly intermediate matched the predicted
evolutionary intermediate. If disassembly is just the reverse of assembly,
this situation is analogous to Haeckel's evolutionary theory:
ontogeny recapitulates phylogeny
(embryonic development resembles the pathway by which the organism evolved).
Additional examples (experimental data from the literature):
I didn't understand what they said about counting face-to-back interfaces
twice, so I looked at examples.