
Usage:
nucleotides atom-spec
mode mode-options
The nucleotides command generates special representations of
nucleic acids, with choices for mode:
- atoms
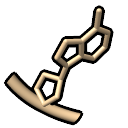 – atomic representation, no special representations; undo the other modes
(same as ~nucleotides)
– atomic representation, no special representations; undo the other modes
(same as ~nucleotides)
- fill
 – atomic representation, but with filled rings
– atomic representation, but with filled rings
- ladder
 – continuous or broken rungs depending on
hydrogen bond pseudobonds
– continuous or broken rungs depending on
hydrogen bond pseudobonds
- stubs
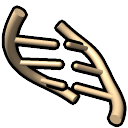 – broken rungs
– broken rungs
- tube/slab


 – tube representation of ribose,
slab representation of bases (box, muffler, or ellipsoid shape)
– tube representation of ribose,
slab representation of bases (box, muffler, or ellipsoid shape)
- slab
 – filled-ring atomic representation of ribose, slab representation of bases
– filled-ring atomic representation of ribose, slab representation of bases
Each mode may have additional options, detailed below.
Nucleotide representations are colored according to the
ring fill color of each residue.
See also:
preset,
color bynucleotide
cartoon,
style,
aniso,
hbonds,
struts,
Actions menu,
Nucleotides icons
•
nucleotides atom-spec
atoms
– or –
•
~nucleotides
atom-spec
Remove special representations to reveal atoms and bonds.
•
nucleotides atom-spec
fill
Atomic representation, but with filled rings (base and ribose).
Fill thickness can be adjusted with style.
•
nucleotides atom-spec
ladder
[ radius r ]
[ showStubs true | false ]
[ baseOnly true | false ]
[ hideAtoms true | false ]
Show nucleic-acid residue pairs connected by
hydrogen bond pseudobonds as rods
or ladder rungs of the specified radius (default 0.45 Å).
Nucleic acid residues without H-bonds to others will be shown as
disconnected half-rungs if showStubs is true (default),
otherwise omitted from the ladder representation.
If baseOnly is true (default),
connecting rungs will only be drawn for H-bonds between base atoms;
if false, thinner connecting rungs (radius 0.2 Å)
will also be drawn for H-bonds involving non-base atoms.
The rungs attach to cartoon at the C3'
tether position, and disconnected half-rungs
(stubs) end at the nucleic acid base N1 (if purine) or N3 (if pyrimidine).
The hideAtoms option indicates whether to hide atoms/bonds
for nucleotides shown as rungs (default true).
Although ladder/stub representions can be limited to specified residues,
the associated parameters are per-model: e.g.,
different residues in the same model cannot have a different rung radius.
•
nucleotides atom-spec
stubs
[ radius r ]
[ hideAtoms true | false ]
Show nucleotide sidechains as disconnected half-rungs of the specified
radius (default 0.45 Å).
The rungs attach to cartoon at the C3'
tether position, and the stubs
end at the nucleic acid base N1 (if purine) or N3 (if pyrimidine).
The hideAtoms option indicates whether to hide the
corresponding atoms/bonds (default true).
•
nucleotides atom-spec
tube/slab
[ radius r ]
[ shape box | muffler | ellipsoid ]
[ dimensions small | long | big | fat ]
[ thickness d ]
[ hideAtoms true | false ]
[ showOrientation true | false ]
[ glycosidic true | false ]
Represent ribose moieties as tubes of the specified radius
(default 0.2 Å) and
bases as slabs of the specified shape, dimensions, and
thickness (default 0.5 Å). The shape can be:
- box (default) – rectangular box
- muffler – rectangular in the plane of the base, oval in cross-section
(large enough to enclose the corresponding box)
- ellipsoid – oval in the plane of the base and oval in cross-section
(large enough to enclose the corresponding box)
The choices for dimensions also specify the location of the slab:
- small (default for ellipsoid shape)
– anchored at the base end of the glycosidic bond
- long (default for box and muffler shapes)
– anchored at the base end of the glycosidic bond
- big – anchored at the ribose end of the glycosidic bond (C1')
- fat – anchored at the ribose end of the glycosidic bond
The hideAtoms option indicates whether to hide nucleotide atoms/bonds
(default true).
The glycosidic option applies only to base-anchored slabs
(dimensions
small or long) and controls whether the ribose tube should
connect to the glycosidic bond (if true) or directly to the slab
(false, default).
The other end of the tube is the average of the C3' and C4' atom positions.
The radius setting applies only to the tube, not the glycosidic bond
that remains visible with glycosidic true;
the stick radius of the bond can be changed separately with the
size command.
The showOrientation option (default false) indicates
whether to show the positive faces of bases with bumps on the slabs.
These faces point towards the 3' end of a strand in right-handed A- and B-DNA,
along the positive Z direction described in:
A standard reference frame for the description of nucleic acid
base-pair geometry.
Olson WK, Bansal M, Burley SK, Dickerson RE, Gerstein M, Harvey SC, Heinemann U, Lu XJ, Neidle S, Shakked Z, Sklenar H, Suzuki M, Tung CS, Westhof E, Wolberger C, Berman HM.
J Mol Biol. 2001 Oct 12;313(1):229-37.
Different residues can have different slab parameters.
•
nucleotides atom-spec
slab
[ shape box | muffler | ellipsoid ]
[ dimensions small | long | big | fat ]
[ thickness d ]
[ hideAtoms true | false ]
[ showOrientation true | false ]
Show ribose as atoms with ring fill and
represent bases as slabs of the specified shape,
dimensions, and thickness (default 0.5 Å),
optionally with showOrientation bumps,
as described above for tube/slab display.
The hideAtoms option indicates whether to hide base atoms/bonds
(default true). Ribose ring-fill can be turned off or its thickness
adjusted with style.
UCSF Resource for Biocomputing, Visualization, and Informatics /
January 2025







