Understanding the mechanisms of cell migration
Investigator: Dyche Mullins, Ph.D.
Institution: University of California, San Francisco
Funding Status: NIGMS R01GM061010 (PI: RD Mullins) May 2000 - April 2015
Significance
Among the most fundamental properties of living cells are the ability to control their shape and the
ability to move. In most eukaryotic cells, shape and movement are driven by assembly of crosslinked
networks of actin filaments in the cytoplasm.
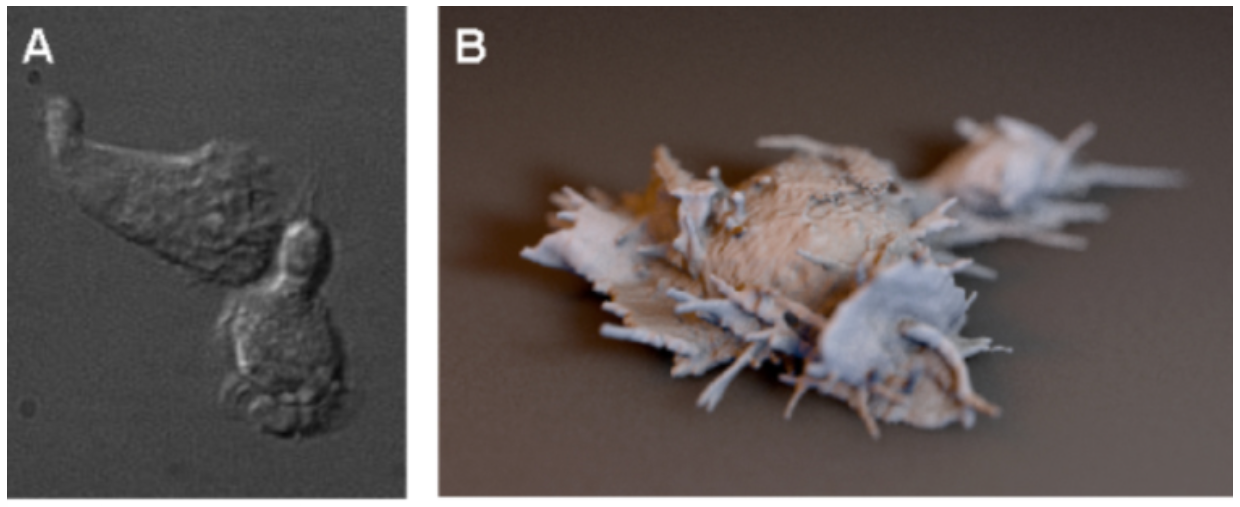
|
|
Figure 1. Light microscopy of neutrophil-like HL60 cells. Left: differential interference
contrast imaging. Right: three dimensional reconstruction of some of our recent Bessel Beam
microscopy data (rendered as an iso-surface contour by UCSF Chimera and shaded by Cinema4D). New
imaging technologies provide dramatic new insights into dynamic cell shape changes driven by actin
assembly.
|
Actin Assembly and Cell Migration.
Despite years of study, the connection between actin
filament assembly and amoeboid cell locomotion remains unclear. This is due, in part, to inherent
molecular and biophysical complexities but it also reflects the fact that cell locomotion is not one
single process. For years the canonical view of migration was that of a single sequence of
coordinated events: (1) actin-driven membrane protrusion; (2) integrin-mediated leading-edge
adhesion; (3) myosin-driven cell body contraction; and (4) force-dependent trailing edge de-adhesion.
Recent work, however, has exploded this simple story and we now realize that eukaryotic
cells use several different mechanisms to crawl. On two-dimensional surfaces most cells depend
heavily on integrin-based adhesions. Crawling through complex, three-dimensional environments,
however, some cells (e.g. fast-moving leukocytes) can move in an integrin-independent manner,
(Lammermann 2008; Lammermann, 2009). The key to this adhesion-independent motility appears to be
spatial confinement. When cells are forced to move through restrictions that are small compared to
the size of their nuclei, weak electrostatic interactions give them purchase required to move
forward (Heuzé, 2013; Renkawitz, 2010).
Dendritic actin networks help drive both two- and three-dimensional cell migration. Loss of the
Arp2/3 complex slows two-dimensional fibroblast migration by 75%, similar to the effect of the actin
polymerization inhibitor, Latrunculin B (Wu, 2012). The residual motility of these cells can still
respond to external chemical cues, but this slow chemotaxis relies on mechanisms of membrane
protrusion that, under normal circumstances, clearly do not contribute much to cell
migration. Interestingly, under certain types of extreme confinement, such as when cells are
squashed between glass coverslips or confined to very narrow channels, some cells can migrate
rapidly in an Arp2/3-independent manner, driven solely by myosin-dependent retrograde flow of
formin-nucleated actin filaments (Renkawitz, 2010; Matthieu Piel, personal communication). In more
complex three-dimensional environments, however, loss of the Arp2/3 complex abolishes dynamic cell
protrusions and dramatically slows migration (Giri, 2013). We are using high-resolution, three-dimensional
light microscopy (Figure 1), mechanical measurements, and biochemically defined mutants of actin
regulators to determine the role of dendritic actin networks in migration of cells through complex
three-dimensional environments.
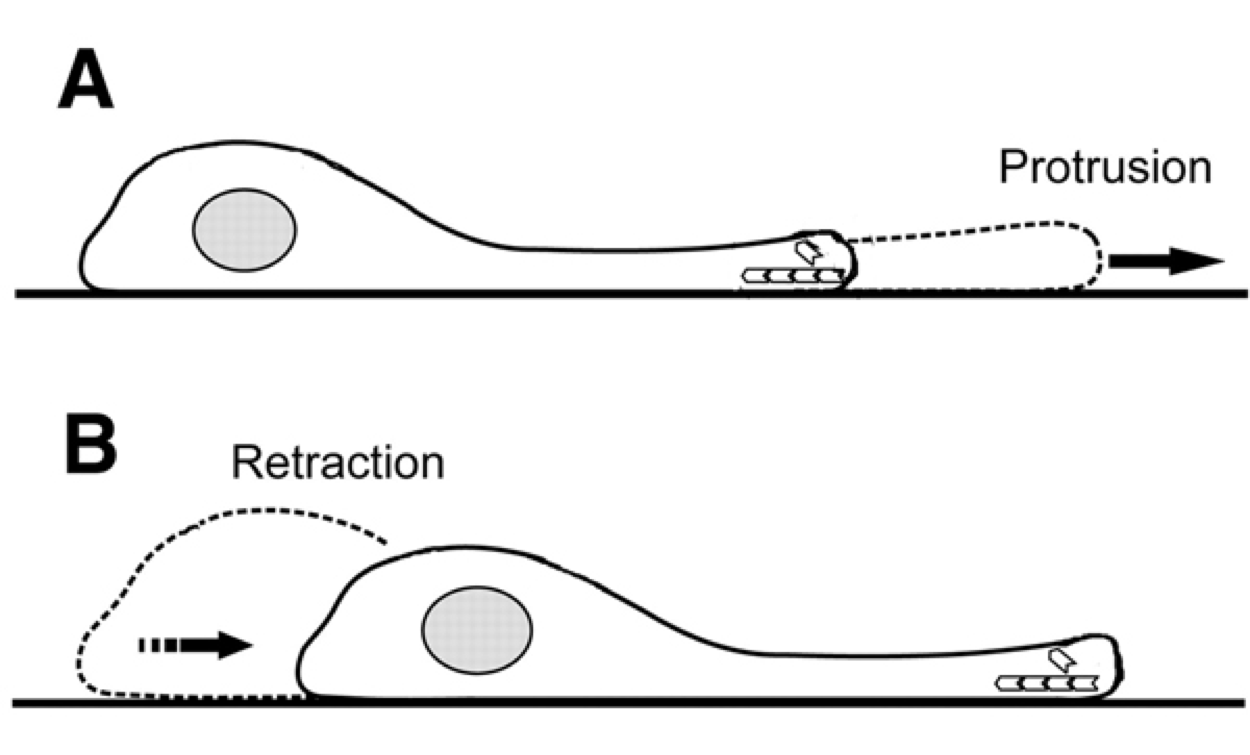
|
|
Figure 2. Protrusion and retraction mechanism for cell motion on two-dimensional
surfaces. Fast three-dimensional microscopy now allows study of the mechanism of motion through
tissues not confined to flat surfaces.
|
Innovation
At least four aspects of this project represent significant innovations: (1) One innovative feature
is the seamless integration across size scales: from single molecule assays, through complex
reconstitutions, to in vivo studies. Compared to studies of other complex cellular structures
(e.g. the mitotic spindle) we have the great advantage of being able to reconstitute the basic
biological function of the lamellipod (generating force and producing movement) from defined
components. This enables us to study regulatory interactions inaccessible in vivo. (2) A second
innovation is the use of three-dimensional Bessel Beam microscopy to follow migration of cells
through complex environments. To extract maximum information we are working with data visualization
specialists (Tom Ferrin and Graham Johnson at UCSF) to create new methods for displaying and
analyzing high-resolution, 3D, time-lapse movies. (3) Thirdly, in collaboration with the Fletcher
Lab at UC Berkeley, we have developed a unique experimental system that enables us to probe
mechanics and composition of functional actin networks in unprecedented ways. (4) Fourthly, we have
developed new tools to visualize actin in nuclei of live cells. Because they are based on
filament-interaction domains of actin binding proteins these probes, unlike GFP-actin, recognize
formin-generated actin filaments.
Decades of work on cell motility has been based on two-dimensional microscopy producing
simple models of protrusion and retraction (Figure 2). Microscope advances have only recently been
able to capture three-dimensional images to characterize cell motion that is not confined to flat surfaces.
Approach
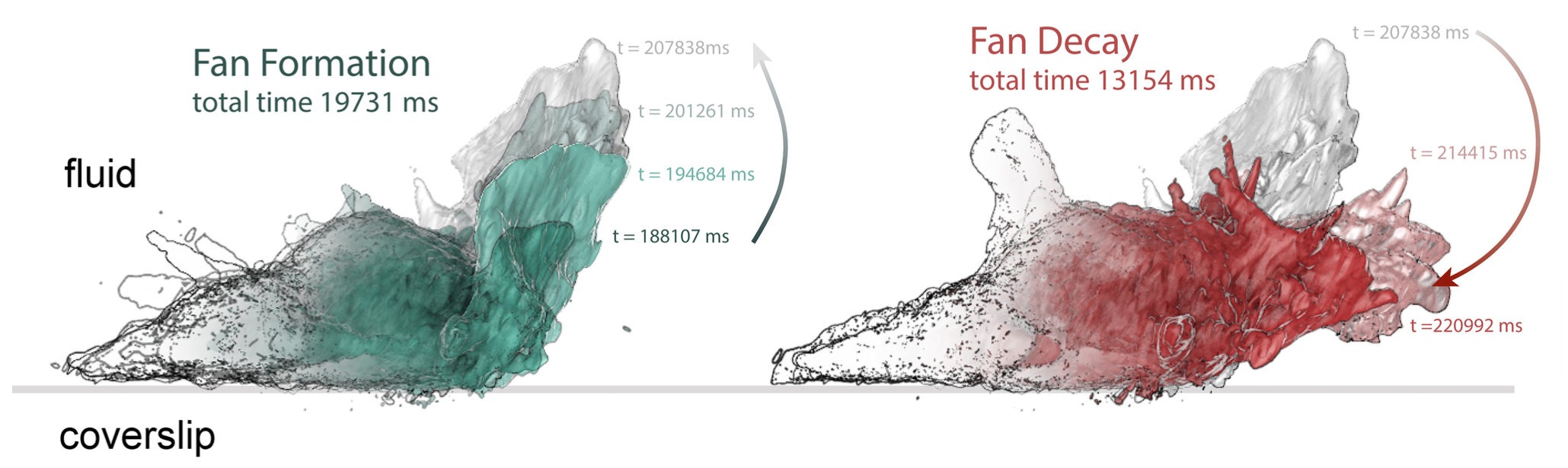
|
|
Figure 3. Life history of a fan- or petal-shaped pseudopod projecting up from the
surface of a neutrophil-like cell crawling on a two-dimensional surface. Three
dimensional data sets from Bessel Beam microscopy have been iso-contour
rendered in Chimera, colorized and overlaid to show extension (left) and collapse
(right) of the pseudopod. The cell is shown in side view.
|
Use high-resolution 3D light microscopy to describe the functional dynamics of membrane protrusions in crawling cells.
Our goal is to understand the fundamental molecular and biophysical bases of rapid,
three-dimensional cell migration. One approach to studying adhesion-independent migration has been
to squeeze cells between passivated coverslips or force them into narrow, microfluidic
channels. This likely mimics movement of some cells through tight spaces, such as extravasion of
neutrophils from the bloodstream, but it does not reproduce conditions experienced by cells
migrating through more complex and compliant matrices. In our studies we will focus on migration of
neutrophil-like cells moving through sparse collagen matrices or microfluidic devices that mimic
normal tissue geometries.
Follow the life-history of three-dimensional "lamellipodial" protrusions in fast-moving
cells.
The morphology and molecular architecture (Iwasa, 2007) of lamellipodial actin networks have
been studied on two-dimensional surfaces but they are not well understood in three dimensions. We
will use Bessel Beam microscopy (Gao, 2014), to characterize membrane protrusions of neutrophils
crawling on flat surfaces and through three dimensional collagen matrices. To analyze complex,
three-dimensional "movies" of locomoting cells we use the open-source data visualization program
UCSF Chimera, developed in Tom Ferrin's laboratory at UCSF (Pettersen, 2004). Chimera began life
as a tool to visualize molecular structures, but recent revisions (Goddard, 2007) enable it to
render density maps generated by three-dimensional light microscopy. The Ferrin laboratory is
currently working with us to further extend Chimera's capabilities to create iso-contour surface
renderings of cells and collagen matrices, extracted from three-dimensional data sets. One goal of
this work is to produce useful three-dimensional analogs of the kymograph or space-time plot (Figure
3), which has proven useful for abstracting information on cellular dynamics from two-dimensional,
time-lapse movies.
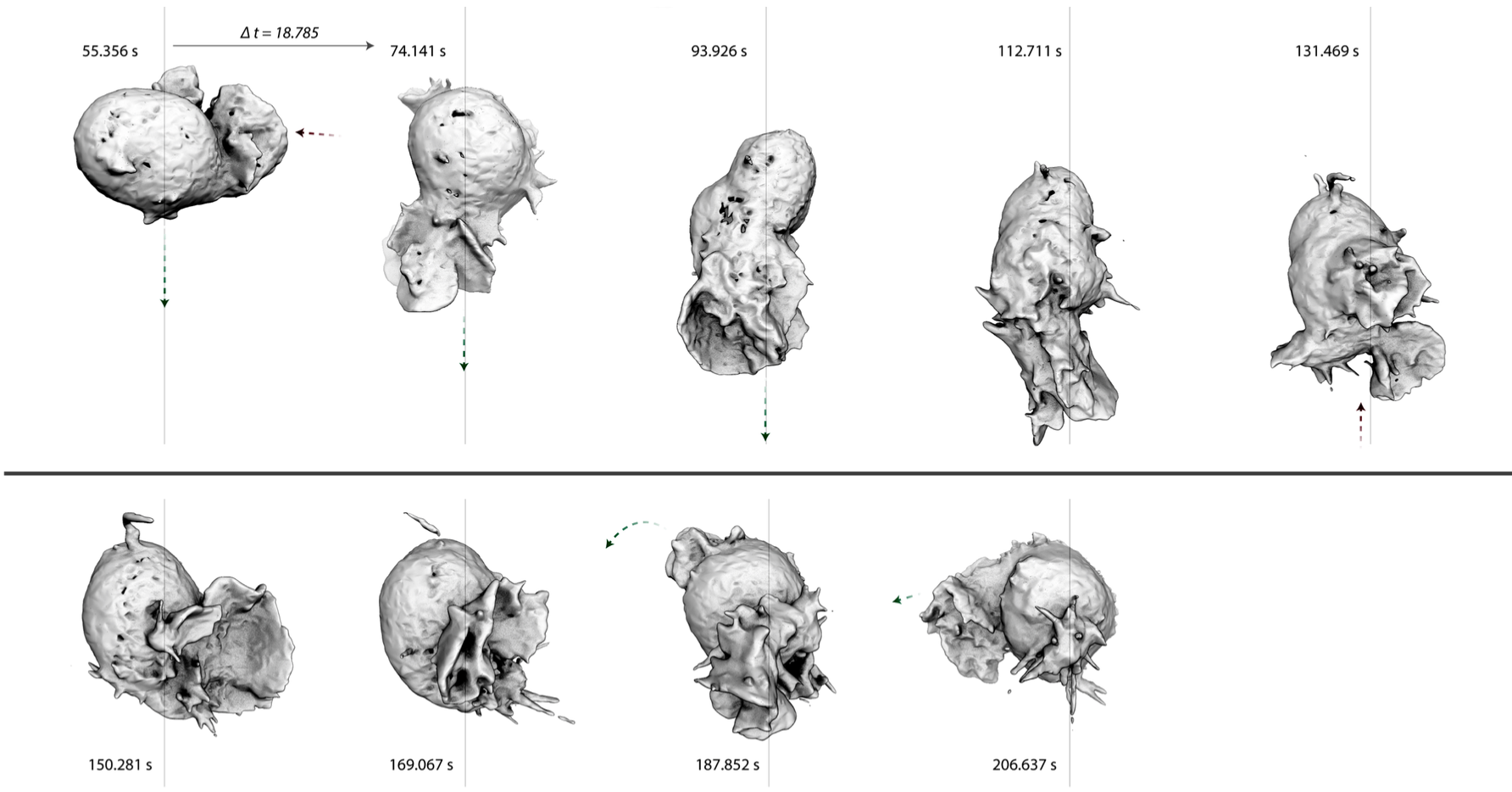
|
|
Figure 4. Iso-contour rendering of Bessel Beam microsocpy of a neutrophil-like cell crawling through
a three-dimensional collagen matrix. Arrows mark the emergence, protrusion, and retraction of
pseudopods.
|
By Bessel Beam microscopy we found that, even when they are not supported by a flat surface,
pseudopodial protrusions are composed of sheet-like "petals" (Figure 4). This is remarkable given
that the dominant model in the field is that planar lamellipodial and lamellar actin networks arise
from strong interactions with flat surfaces (Burnette, 2014). Some pseudopods consist of a single
petal but, in many cases, a protrusion comprises multiple petals, nested to form a rosette. When we
followed their entire life history, we noticed that pseudopods often begin as single, dynamic
filopodia. Similarly, when they disappear, pseudopod rosettes collapse into a jumble of filopodial
spikes. To understand the molecular architecture of these three dimensional pseudopods, we will
perform high-resolution, three-dimensional imaging of the known components of two-dimensional
lamellipodial and lamellar networks: Arp2/3 complex, capping protein, cofilin, and tropomyosin. One
important question is whether the petals have the same architecture as two-dimensional lamellipodia
and lamella (Iwasa, 2007): are they composed of dendritic actin networks sitting on top of
contractile networks of tropomyosin-coated filaments? Does pseudopod collapse represent loss of the
dendritic network or contraction of an underlying network? Also, do the residual filopodia that
remain after collapse of the rosettes represent structures that were present the entire time? Does
each lamellar petal have a filopodium at its heart? Also, do these filopodia contain Ena/VASP- or
formin-family proteins? If we find that the protrusion of petals is always proceeded by filopodia,
that would strongly suggest that pseudopod generation is a multi-step process, with a filopodial
initiation phase and a stable, lamellar growth phase. Such a multi-step mechanism might explain
several general features of cell migration, including the effect of membrane tension on the
outgrowth of new pseudopods (Houk, 2012).
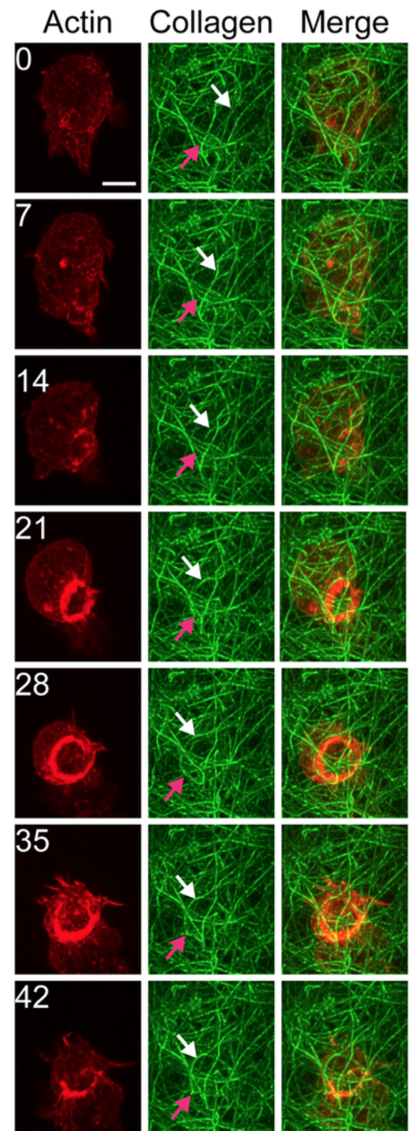
|
|
Figure 5. Time-lapse Bessel Beam microscopy of neutrophil-like HL-60 cell crawling through a
three-dimensional collagen matrix. Red: filamentous actin labeled by mCherry-utrophin-260. Green:
fluorescein collagen. Arrows indicate collagen fibers displaced by passage of the cell. The cell
assembles a massive actin ring as it passes through a constriction.
|
Characterize the interaction of three-dimensional lamellipodial "petals" with the extracellular
environment.
Forces generated by cells crawling on two dimensional surfaces have been measured many
times (Plotnikov, 2014) and always turn out to be contractile. The distribution of forces around
cells crawling in three dimensions have never been carefully measured. We aim to
determine the direction and relative magnitude of the forces applied by fast-moving cells to the
extracellular matrix. Briefly, we will image neutrophil-differentiated HL-60 cells, expressing a
membrane-targeted mCherry, as they move through fluorescein-labeled collagen fibers. We will extract
the network architecture of the collagen matrix from every frame of the movie using "Network
Extractor" and "Image Surfer" (Feng, 2007), programs written especially for characterizing
three-dimensional collagen matrices. After extracting the network architecture we will identify
vertices and midpoints of all the network segments. We will then analyze the movements of the
segment midpoints as the cell passes through the network. We will normalize the displacement by the
thickness of the fiber so that relative displacement corresponds to relative force. Our preliminary
data reveal that, in contrast to two-dimensional cell migration, neutrophils exert almost no
pulling forces as they pass through a collagen matrix. Almost all of the forces appear to be pushing
outward, away from the membrane. Also, when cells reach a constriction in the collagen matrix they
generally polymerize a significant amount of actin in contact with the collagen, leading to very
large, outward deformation of the collagen fibers around the cell (Figure 5). We will also
correlate the pushing forces with local cell morphology. For example, in what direction do the
forces around the cell body point? What types of forces are transmitted by growing and ruffling
lamellar petals?
Compare the migration and membrane dynamics of normal and perturbed cells.
We intend to couple tools and insights developed from methods of the previous two sections with pharmacological and
genetic perturbations to work out the molecular mechanisms underlying three-dimensional membrane
protrusion and cell migration. We will, for example, compare the morphology and life cycle of
membrane protrusions generated by: (i) normal cells; (ii) cells treated with cytoskeletal
inhibitors; (iii) cells in which expression of actin regulators has been knocked down; and (iv)
cells expressing biochemically defined mutant versions of actin regulatory proteins. Briefly, we
will determine the source of the actin filaments (Arp2/3 complex, formins, etc.) generated at sites
of intimate contact with constrictions in the collagen matrix. We will
determine the roles of WASP and WAVE in three-dimensional cell migration and determine the extent of
crosstalk between these nucleation promoting factors.
We will, for example, knock down WASP and WAVE expression and characterize the morphology of the
cells as well as their mechanical coupling to the collagen matrix. This is an extremely important
experiment as we hypothesize that cells employ different biophysical mechanisms to carry out three
dimensional migration in the absence of WASP and WAVE. We will knock down WAVE
expression and rescue cells with WAVE truncations to determine whether the capacity to activate the
Arp2/3 complex is essential for WAVE's role in pseudopod formation and cell migration.
References
Burnette DT, Shao L, Ott C, Pasapera AM, Fischer RS, Baird MA, Der Loughian C, Delanoe-Ayari H, Paszek MJ, Davidson MW, Betzig E, Lippincott-Schwartz J. (2014) A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells. J Cell Biol. 205(1):83-96.
Feng D, Marshburn D, Jen D, Weinberg RJ, Taylor RM 2nd, Burette A. (2007) Stepping into the third dimension. J Neurosci. 27(47):12757-12760.
Gao L, Shao L, Chen BC, Betzig E. (2014) 3D live fluorescence imaging of cellular dynamics using Bessel beam plane illumination microscopy. Nat Protoc. 9(5):1083-1101.
Giri A, Bajpai S, Trenton N, Jayatilaka H, Longmore GD, Wirtz D. (2013). The Arp2/3 complex mediates multigeneration dendritic protrusions for efficient 3-dimensional cancer cell migration. FASEB Journal 27(10):4089-4099.
Goddard TD, Huang CC, Ferrin TE. (2007) Visualizing density maps with UCSF Chimera. J Struct Biol. 157(1):281-287.
Heuze ML, Vargas P, Chabaud M, Le Berre M, Liu Y-J, Collin, O., et al. (2013). Migration of dendritic cells: physical principles, molecular mechanisms, and functional implications. Immunol. Rev., 256(1):240-254.
Houk AR, Jilkine A, Mejean CO, Boltyanskiy R, Dufresne ER, Angenent SB, Altschuler SJ, Wu LF, Weiner OD. (2012) Membrane tension maintains cell polarity by confining signals to the leading edge during neutrophil migration. Cell. 148(1-2):175-188.
Iwasa JH, Mullins RD. (2007) Spatial and temporal relationships between actin-filament nucleation, capping, and disassembly. Curr Biol. 17(5):395-406.
Lammermann T, Sixt M. (2009) Mechanical modes of 'amoeboid' cell migration. Curr Opin Cell Biol. 21(5):636-44.
Lammermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Soldner R, Hirsch K, Keller M, Förster R, Critchley DR, Fassler R, Sixt M. (2008) Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 453(7191):51-5.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 25(13):1605-12.
Plotnikov SV, Sabass B, Schwarz US, Waterman CM. (2014) High-resolution traction force microscopy. Methods Cell Biol.123:367-394.
Renkawitz J, Sixt M. (2010). Mechanisms of force generation and force transmission during interstitial leukocyte migration. EMBO Reports, 11(10):744-750.
Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, et al. (2012). Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell, 148(5):973-987.