This tutorial is an introduction to working with sequence alignments and associated structures using Multalign Viewer. Data are taken from the enolase superfamily:
The enolase superfamily: a general strategy for enzyme-catalyzed abstraction of the alpha-protons of carboxylic acids. Babbitt PC, Hasson MS, Wedekind JE, Palmer DR, Barrett WC, Reed GH, Rayment I, Ringe D, Kenyon GL, Gerlt JA. Biochemistry. 1996 Dec 24;35(51):16489-501.To follow along, first download the sequence alignment file super8.msf to a convenient location. This sequence alignment contains the barrel domains of eight enolase superfamily members.
Start Chimera by clicking or doubleclicking the Chimera icon
![]() (depending on its location).
Typically, this icon will be present on the desktop.
The Chimera executable can also be run from its
installation location (details...).
(depending on its location).
Typically, this icon will be present on the desktop.
The Chimera executable can also be run from its
installation location (details...).
A splash screen will appear, to be replaced in a few seconds by the main Chimera graphics window or Rapid Access interface (it does not matter which, the following instructions will work with either).
Choose File... Open, then locate and open super8.msf. Opening a sequence alignment file automatically starts Multalign Viewer and uses it to display the alignment.
| Multalign Viewer sequence window |
|---|
 |
Multalign Viewer has its own set of preferences, including display options. From the alignment window menu, choose Preferences... Appearance and adjust settings as desired for Multiple alignments, then Close the preferences dialog.
Size and place the sequence window and main Chimera window so that both are visible. If the sequence window later becomes obscured, it can be raised by choosing Tools... MAV - super8.msf... Raise from the menu, or by clicking super8.msf in the lefthand Active Dialogs section of Rapid Access (shown/hidden by clicking the lightning-bolt icon at the bottom of the Chimera window).
A Consensus sequence and Conservation histogram are shown above the sequences. They can be hidden/shown using the Headers menu. Red capital letters in the consensus sequence indicate a few positions in which all of the sequences have the same type of residue.
The first sequence in the alignment, named mr, corresponds to the barrel domain of mandelate racemase from Pseudomonas putida. The next-to-the last sequence in the alignment, named enolyeast, corresponds to the barrel domain of enolase from Saccharomyces cerevisiae. There are multiple structures in the Protein Data Bank for each of these sequences; 2mnr (mandelate racemase) and 4enl (enolase) are used in this tutorial.
If you have internet connectivity, structures can be obtained directly from the Protein Data Bank. Use Fetch by ID (File... Fetch by ID in the Chimera menu) to get 2mnr and then 4enl from the PDB database. If you do not have internet connectivity, instead download the files 2mnr.pdb and 4enl.pdb included with this tutorial and open them in that order with File... Open.
The view is initially centered on the first structure opened, mandelate racemase. Off to the side and possibly out of view is the second structure, enolase. Use the menu to bring everything within view:
Actions... FocusApply the ribbons preset (which may or may not change the appearance, depending on your preference settings):
Presets... Interactive 1 (ribbons)Move and scale the structures as desired throughout the tutorial.
| sequence window after structure association |
|---|
 |
Notice that in the sequence window, the sequence names mr and enolyeast are now shown in bold within colored boxes. Each structure was automatically associated with its best-matching sequence, and the colors indicate the pairing.
Association enables several types of crosstalk, which will be explored in more detail below:
First, we will use the sequence alignment to guide a structural match. From the Multalign Viewer menu, choose Structure... Match and click Apply without checking any boxes. This superimposes the structures using the alpha-carbons of all pairs of residues aligned in the sequence alignment. Readjust the view to focus on the active sites, in this case, the metal ions and surrounding atoms:
Select... Structure... ionsClear the selection by Ctrl-clicking in an empty part of the graphics window.
Select... Zone... (try an 8-Å cutoff)
Actions... Focus
Match statistics are reported briefly in the status line at the bottom of the Chimera window and written to the Reply Log (Favorites... Reply Log). The match is fairly rough, giving an RMSD of 8.4 Å, and the active sites are somewhat offset. Apparently, not all 175 pairs of residues in the same columns of the sequence alignment superimpose well in space. Try matching again, but this time, turn on the option to Iterate by pruning... and edit the angstrom value to 1.0 before clicking OK. This improves the superposition of the active sites by omitting dissimilar parts from the fit.
| active sites |
|---|
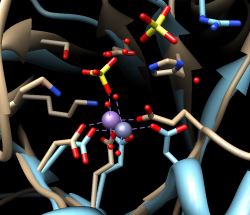 |
Zoom back out to view the overall structures:
Actions... FocusThe structures have N-terminal and barrel domains, but only the barrel domains are included in the sequence alignment. With the mouse, drag a box to highlight the entire sequence alignment. That will select the corresponding parts of the proteins. Invert the selection to the parts of the proteins not included in the sequence alignment, then hide the ribbon for those parts:
Select... Invert (all models)Boxes drawn on the alignment are called regions. A single region may consist of several disconnected boxes. The active region is shown with a dashed outline. Delete the active region by clicking in some blank area of the sequence window (not on the sequences) to make sure this window has the mouse focus, then pressing the keyboard delete key. If the region is not active (the border is not dashed), first click on the region to activate it, then press delete.
Actions... Ribbon... hide
Select... Clear Selection
Actions... Focus
Communication also goes in the opposite direction: a selection in the structures is shown on the sequence alignment as a region. For example, select all aromatic residues in the structures:
Select... Residue... amino acid category... aromaticThe green boxes in the sequence alignment are the selection region. Clear the selection by Ctrl-clicking in an empty part of the graphics window.
Placing the cursor over any structure-associated residue in the sequence alignment shows the corresponding structure residue number near the bottom of the sequence window. For example, clicking into the sequence window and then placing the cursor over the first residue in the mr sequence shows that it is associated with Val 134 in chain A of model #0 (2mnr, mandelate racemase).
Next, see where some of the conserved residues are within the structures. In the sequence window, find the first completely conserved residue in the alignment. It is an aspartic acid, D, at alignment position 99. Drag with the mouse to create a region containing just that column of the alignment. The corresponding structure residues will be selected.
These aspartic acid residues are already displayed because the ribbons preset (used above) automatically displays sidechains near any bound ions or ligand molecules.
At alignment position 150, there is a completely conserved proline (P). Create another region for this column (this time, press Ctrl along with the mouse button to start a new region instead of replacing the first). Only ribbons are shown for these residues; display the sidechain atoms too:
Actions... Atoms/Bonds... showAlignment regions can also be created automatically. From the Multalign Viewer menu, choose Structure... Secondary Structure... show actual. This creates regions in the structure-associated sequences named structure helices (light yellow with gold outline) and structure strands (light green with green outline).
The region names are listed in the Region Browser (Info... Region Browser in the Multalign Viewer menu). Clicking the Active checkbox for a region will select the corresponding residues in any associated structures. Only one region can be active at a time. Close the Region Browser.
| conservation shown with color |
|---|
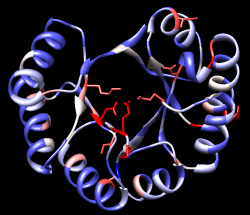 |
Choose Tools... General Controls... Command Line from the main Chimera menu and use a command to close the enolase structure:
Command: close 1Clear the selection (if any) by Ctrl-clicking in an empty part of the graphics window.
A structure can be colored according to the conservation in an associated sequence alignment. Choose Structure... Render by Conservation from the Multalign Viewer menu. The resulting Render by Attribute tool shows a histogram of the residue attribute named mavConservation. The “mav” part of the name is shorthand for Multalign Viewer, and “Conservation” indicates the values correspond to what is shown in the Conservation line of the sequence window. By default, the value for a column is the fraction of sequences with the most common residue type at that position, for example: 1.0 where all 8 sequences have the same type of residue, and 2/8 = 0.25 where only 2 (but any 2) share a type.
The Render by Attribute histogram contains vertical bars (or thresholds) for mapping the attribute values to colors, radii, or worms. Use the Colors section. You can define your own color mapping by dragging the thresholds along the histogram and/or changing their colors. The Value and Color are shown for the most recently clicked or moved threshold. The Value changes when the threshold is moved, or the position can be changed by entering a value and pressing return. The Color can be changed by clicking the square color well and using the Color Editor. Adjust the color mapping as desired before clicking Apply. Coloring the structure shows the high conservation of the metal-binding residues and the low conservation of most residues around the outside of the barrel.
Attributes can also be used in the command line. For example, show only the residues with mavConservation values greater than 0.7:
Command: show :/mavConservation>.7The conservation can be calculated in different ways (entropy, variability, etc.) using the AL2CO program included with Chimera. Choose Preferences... Headers from the Multalign Viewer menu, set Conservation style to AL2CO, and adjust additional AL2CO parameters as desired. The Conservation line in the sequence window will update automatically. In Render by Attribute, it may be necessary to use Refresh... Values to update the histogram before recoloring the structure.
Multalign Viewer includes many features, only a few of which are sampled here. Users are encouraged to explore the menus and documentation for more information. See also the Mapping Sequence Conservation tutorial on the Chimera website.
End the Chimera session:
Command: stop now