Unit Cell 
Unit Cell builds crystallographic unit cells
using symmetry information from the input coordinate file (PDB or mmCIF).
A crystallographic unit cell consists of a unique set of coordinates,
duplicated and transformed according to the crystallographic and
(if present) noncrystallographic symmetries in the crystal.
Unit Cell can be used to regenerate the full unit cell, or only
those parts defined by
crystallographic symmetry or noncrystallographic symmetry.
See also:
Crystal Contacts,
Multiscale
Models,
sym,
the biological
unit function in the
Model Panel,
fetching PDB-biounit and PQS files
There are several ways to start
Unit Cell, a tool in the Higher-Order Structure category.
After a PDB or mmCIF file has been
opened in Chimera,
Unit Cell can generate symmetry-related
copies if the file contains sufficient information.
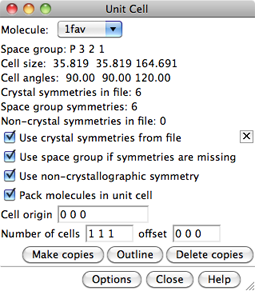
In the dialog, Molecule can be set to any open molecule model.
The space group, unit cell parameters, and numbers of transformation matrices
for the current Molecule are shown, if available.
- Make copies loads copies of the coordinates from the
same source as the originally opened model (a local file or a file
fetched from the Web) and transforms them as
specified by the options.
- Outline toggles whether a white outline of the unit cell is shown.
- Delete copies removes all copies of the molecule except the original.
The copies can be saved.
Close simply dismisses the dialog, while Help opens this
page in a browser window.
Clicking Options reveals additional settings
(clicking the close button  on the right
hides them again).
The descriptions below refer to PDB files,
but the corresponding mmCIF information can also be used:
on the right
hides them again).
The descriptions below refer to PDB files,
but the corresponding mmCIF information can also be used:
- Use crystal symmetries from file
- apply crystallographic symmetry described by
SMTRY1, SMTRY2, and SMTRY3 matrices in REMARK 290 lines of a PDB file
- Use space group if symmetries are missing
- for files without crystallographic symmetry matrices,
use the space group name in the CRYST1 record of a PDB file
to look up the crystallographic symmetry. Space group names are the full
Hermann-Mauguin symbols, e.g., P 1 21 1 rather than P 21
(see lists of names).
- Use non-crystallographic symmetry
- apply noncrystallographic symmetry described by
MTRIX1, MTRIX2, and MTRIX3 matrices in a PDB file. Only a small fraction of
PDB entries have MTRIX records (~10% in 2003). Most of these describe how
to transform the coordinates of one chain to match closely the coordinates
of another chain already present in the PDB file. In a few cases (~0.25%
of PDB entries), the MTRIX records describe a transformation that produces
a new copy whose coordinates are not already in the file.
These two cases are distinguished by the contents of
column 60 in the MTRIX records. A "1" means that
the transformation yields coordinates that are already given (these
MTRIX records are ignored by Unit Cell), whereas a blank space
means that the transformation will produce a new copy.
MTRIX column 60 may be set incorrectly in some files. Further,
MTRIX records are sometimes missing. For example, 1cd3 has no MTRIX
records, but the remarks describe how to produce them from the BIOMT
records. Of course, this cannot be handled by Unit Cell.
MTRIX records do not describe crystallographic symmetries, but
additional symmetries of the asymmetric unit of the crystal.
Because the copies of the molecule occupy nonequivalent positions
in the crystal, they usually have small structural differences due
to differing crystal contacts.
Thus, an independent set of coordinates is usually included for these copies,
and MTRIX records are not needed.
MTRIX records are often used for icosahedral virus particles, where
the PDB file will include the coordinates of one molecule in the shell
and MTRIX records describing how to place copies that are
not crystallographically equivalent.
The size of the virus particle precludes independent refinement
of coordinates for each copy of a molecule in the shell.
- Pack molecules in unit cell
- whether to pack the chains so that their centers fall within
one unit cell box
- Cell origin coordinates (default 0 0 0)
are in unit cell lengths and describe translations of the unit cell
along its axes.
Values of a coordinate that differ by integer amounts are equivalent;
e.g., (0.3 0 0) is equivalent to (-0.7 0 0) and (4.3 0 0).
Changing the origin may rearrange the copies and shift the outline (if present).
The shift will occur in the direction that maintains the center
of the original copy within the box.
- Number of cells (default 1 1 1, a single cell)
offset (default 0 0 0) allows
generating a block of multiple unit cells.
The first three values specify dimensions in number of cells
along the unit cell axes; the second three values specify the placement
of the block relative to the cell containing the original structure.
For example, number of cells 3 1 1 and offset –1 0 0
would give a 3x1x1 block of cells with the original structure
in the middle cell along the first axis.
Changes in cell origin or number can be applied by pressing Enter (return)
or clicking the Make copies button.
Limitations
Many problems are due to information that is missing
from (or incorrect within) the input file.
One way to validate symmetry information is to identify steric clashes
with the Crystal
Contacts tool.
Unit Cell does not generate multimers defined by
BIOMT matrices,
but this can be done with
Multiscale
Models or the command
sym.
Only the following CIF unit cell fields are used:
symmetry_space_group_name_h-m
symmetry_equiv_pos
symmetry_equiv_pos_site_id
symmetry_equiv_pos_as_xyz
space_group_symop_operation_xyz
cell_length_a, cell_length_b, cell_length_c
cell_angle_alpha, cell_angle_beta, cell_angle_gamma
cell_formula_units_z
UCSF Computer Graphics Laboratory / June 2014