| 3hj2 Glu16 rotamers (see list) |
|---|
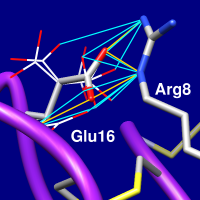 |
The Rotamers tool allows amino acid sidechain rotamers to be viewed, evaluated, and incorporated into structures. A residue can be changed into a different rotamer of the same type of amino acid or mutated into a different type. The state of Rotamers is included in saved sessions. See also: Adjust Torsions, Dock Prep, Ramachandran Plot
Although sidechains at multiple positions can be replaced simultaneously, the Rotamers tool is not recommended for predicting the conformations of multiple sidechains in an interacting cluster. Programs such as SCWRL are more appropriate for that purpose.
There are several ways to start Rotamers, a tool in the Structure Editing category. Rotamers is also implemented as the command swapaa.
One or more amino acid residues must be selected to indicate where the rotamers should be placed. Partly selected residues will be considered selected, and if more than one residue is selected, a rotamer set (of the same residue type) will be placed at each.
In the Dynameomics rotamer library only, there are multiple choices of type for cysteine and histidine depending on the oxidation or protonation state of the sidechain:
For nonstandard amino acids, see the SwissSidechain Chimera plugin.
A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Shapovalov MV, Dunbrack RL Jr. Structure. 2011 Jun 8;19(6):844-58.
The Dynameomics rotamer library: amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water. Scouras AD, Daggett V. Protein Sci. 2011 Feb;20(2):341-52.
The penultimate rotamer library. Lovell SC, Word JM, Richardson JS, Richardson DC. Proteins. 2000 Aug 15;40(3):389-408.
Close dismisses the Rotamers dialog, while Help opens this manual page in a browser window.
When Apply (or OK, which also dismisses the dialog) is clicked, phi and psi angles at each selected position are measured and reported in the Reply Log. If a type without a rotamer set (glycine, alanine) has been specified, the residue(s) will simply be changed to the new type, and the change cannot be undone. Otherwise, for each selected position, the specified rotamer set will be displayed and a rotamer list will appear. Bond lengths and angles are taken from the Amber parameter files all*94.lib, and hydrogens are not included. The rotamers are placed on the existing residue by superimposing the CA, CB, and C atoms, or if the existing residue does not have a CB atom, the N, CA, and C atoms.
| rotamer list (see display) |
|---|
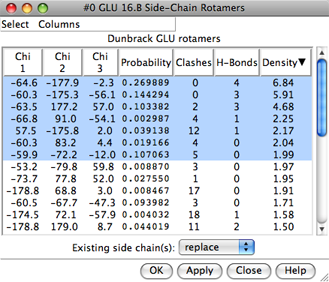 |
Each rotamer list includes sidechain torsion (Chi) and Probability values. Probabilities are simply taken from the rotamer library and are not affected by the structural environment, except by phi and psi angles when the Dunbrack library is used. Rotamers are initially listed in order of decreasing probability, but the listing can be sorted by the values in any column by clicking the header. Clicking the header once sorts the entries in order of increasing value and places an up arrowhead in the header. Clicking again sorts the entries in decreasing order and places a down arrowhead in the header.
The rotamer-list Columns menu controls which columns are shown and allows rotamers to be evaluated in terms of clashes or contacts, hydrogen bonds, and/or overlap with a density map. The rotamer-list Select menu allows rotamers to be selected for subsequent operations such as with the Actions menu or commands.
One or more rotamers can be chosen from the list by clicking and dragging with the left mouse button; Ctrl-click toggles whether a rotamer is chosen. Only the chosen rotamers are displayed.
Clicking Apply (or OK, which also dismisses the list) will incorporate the chosen rotamer(s). When there will be multiple sidechains at a given residue position, the new sidechain(s) will be assigned different alternative location identifiers. The Existing side chain(s) setting controls what happens to the pre-existing sidechains. They can be replaced, retained, or selectively retained based on selection. After a sidechain has been replaced, it cannot be recovered except by reopening the structure. The display style of the former sidechain will be used. Other characteristics, however, will not be transferred and may need to be regenerated (for example, with AddH and Add Charge). Close dismisses the list and deletes the set of rotamers without further modifying the structure.
Rotamers can be evaluated in the context of the structural environment or compared to density (or other volume data). Only the sidechain atoms of a rotamer are evaluated. For hydrogen bond and clash detection, interactions with rotamers in the same set and the current residue at that position are disregarded, but all other atoms in the vicinity will be included unless Ignore... other models (see below) is used. That setting is useful for preventing superimposed related proteins or additional copies of the starting structure from contributing to the counts. In addition, atoms in the same model that are unwanted for such calculations (for example, solvent) should be deleted beforehand.
Columns... Add... Clashes brings up a dialog for finding clashes or contacts and showing the results in the rotamer list. This uses capabilities from Find Clashes/Contacts:
The overlap between two atoms is defined as the sum of their VDW radii minus the distance between them and minus an allowance for potentially hydrogen-bonded pairs:overlapij = rVDWi + rVDWj – dij – allowanceijAn allowance > 0 reflects the observation that atoms sharing a hydrogen bond can come closer to each other than would be expected from their VDW radii. It is only subtracted for pairs comprised of a possible hydrogen-bond donor atom (or donor-borne hydrogen) and possible acceptor atom.The Default criteria are appropriate for clash detection. For detecting clashes, cutoff values of 0.4-1.0 Å and allowance values of 0.2-0.6 Å are generally reasonable. For detecting contacts, negative cutoff values of 0.0-(–1.0) Å with an allowance of 0.0 Å are generally reasonable.
Clicking Apply (or OK, which also dismisses the dialog) performs the calculation and shows the results in the rotamer list. Close simply dismisses the dialog, while Help opens this manual page in a browser window.