Publication Images and Movies Course
July 29, 2008
UCSF Mission Bay Library
Data and scripts. Data and script files needed for this tutorial (0.1 Mbytes): movie-data.zip
Chimera version. This tutorial was tested with Chimera version 1.2540 (July 9, 2008). It may not work with older Chimera versions.
Mac X Server settings. To avoid clicking twice on Chimera dialogs on the Mac with X windows (once to activate window, second time to press a button) type the following command to a Mac Terminal and restart X11.
defaults write com.apple.x11 wm_click_through -bool true
Mac copy and paste. To paste a copied command to the Chimera command-line in the Mac X11 version of Chimera use the middle mouse button. The Mac Aqua (native windows) Chimera uses the usual Mac copy and paste (Apple+c, Apple+v).
MPEG license. The first time you write a Chimera movie using MPEG-2 or MPEG-4 encodings you'll be asked to agree to the MPEG license agreement.
|
|
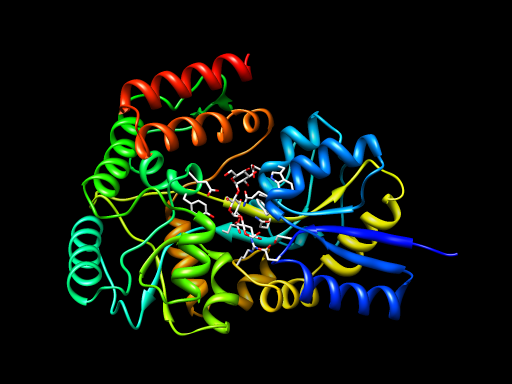
Simplest kind of movie -- rotate scene 360 degrees.
Setup scene: open 2gha-chain-A.pdb, use Preset / Interactive ribbons, select ligand atom and set pivot. Then open Chimera command file spin.cmd containing the following commands.
movie record turn y 3 120 wait 120 movie stop movie encode output spin.mov
The quicktime movie file appears in the same directory as the spin.cmd script. View it.
Show the binding pocket by zooming in by hand.
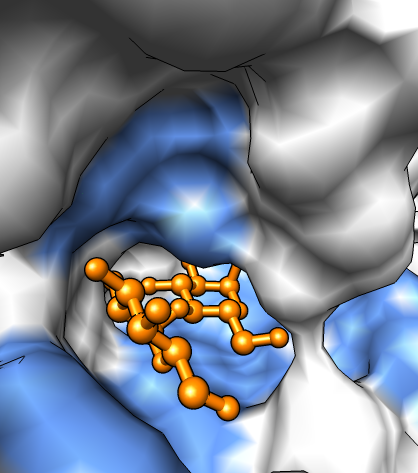
|
First set up the scene.
You could use lots of other image enhancement tricks: silhouette edges on white background for sense of depth, subdivision quality for smoother atoms and bonds, vertex density for smoother surface, shininess adjustment....
Practice movie, rock model left-right, zoom in, rock left-right.
Show Movie Recorder dialog, Tools / Utilities / Movie Recorder. Press Record and go through hand motion, rock, zoom, rock. Press Stop. A few hundred frames have been captured.
Want to show more depth in pocket. We will record some more frames to show this. Show side view. Set view to top. Press Record. Widen field of view with side view red lines, rock model, press Stop.
Encode the movie. Press Browse to choose file name pocket.mov on desktop. Press Make Movie. View movie.
Smoother motion can be obtained using a command script with the rock and scale commands (pocket.cmd). Limitation: There is no command to adjust field of view. That could be done in a difficult way with a Python script.
Now we'll show how to morph between two conformations of maltotriose binding protein, the bound and unbound states.
Setup the scene: open the unbound conformation 2ghb-chain-A.pdb, close surface, use Preset / interactive ribbons.
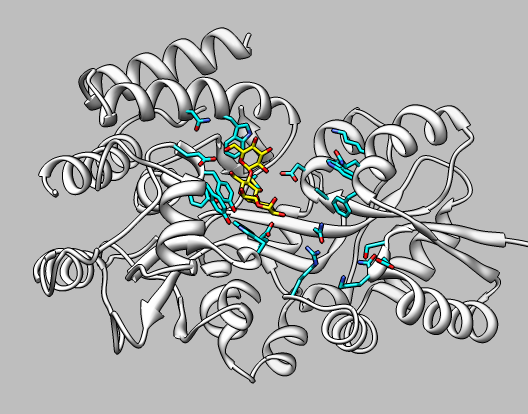
|
Morph: Start Morph Conformations (menu Tools / Structure Comparison), use Add... button to add unbound state (2ghb), followed by bound state (2gha), and finishing with unbound state (2gha) (this makes the morph loop back to the start). Press Create. Takes a minute to calculate morph.
Hide original models. Press play button on MD Movie player.
Put maltotriose ligand in: show 2gha, select ribbon residue with mouse, up arrow key, delete atoms.
Put in nearby residues: zone (whole residues), show atoms.
Color ribbons gray, color ligand yellow with model panel color color button.
Orient with ligand at top using frame 21 (bound state) and play trajectory.
Record movie (MD Movie menu File / Record Movie...) using frame quality "screen" for faster recording, and additional encoding options "bitrate 6000" for better movie quality. In quicktime player can use View / Loop menu entry to play in a loop.
This highlights the ligand by showing it in motion travelling to its binding site.
Save three positions: 1) MBP centered and ligand bound, 2) MBP in lower right corner and ligand bound, 3) MBP in lower left corner and ligand in upper right corner. Use the savepos command and name these closeview, wideview, and farligand. Use the Model Panel active button to move the ligand to create position 3.
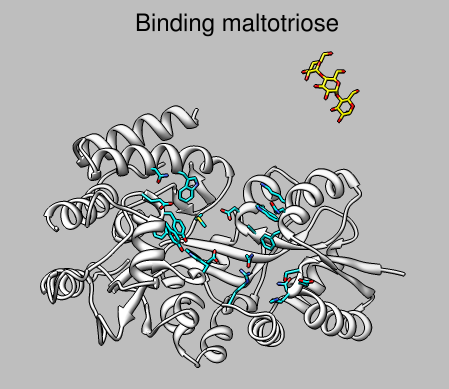
|
Try these commands to replay motion 3 -> 2 -> 1.
reset farligand reset wideview 50 reset closeview 25
Open the command script fly.cmd to record a movie of this. The movie file fly.mov will be created in the same directory as the fly.cmd script.
The command script also fades in and out a text label.
reset farligand 2dlabel create title text "Binding maltotriose" 2dlabel change title xpos 0.3 ypos 0.92 visibility hide movie record 2dlabel change title visibility show frames 25 wait 25 reset wideview 50 wait 50 reset closeview 25 2dlabel change title visibility hide frames 25 wait 25 movie stop movie encode output fly.mov bitrate 6000
|
We'll try to string together the above steps: first the ligand flies in to the unbound state of the protein, then the protein morphs to the bound state, then we show a close-up view of the binding site, add a surface, and color the aromatic residues on the surface. We'll also show hydrogen bonds between the ligand and protein after the morph finishes.
Our strategy will be to write a command script that creates the entire movie. This makes it easy to change the movie without repeating lots of manipulations with dialogs and menus.
Try all the commands interactively. The purpose of the exercise is to get familiar with the commands.
Additional information about commands is found in the Chimera User's Guide.
Restart Chimera. Show the Command-line (use Favorites menu).
Change the directory to the one that contains the PDB files and open the bound and unbound conformations.
cd ~/Desktop/movie-data open 2gha-chain-A.pdb open 2ghb-chain-A.pdb
Use interactive preset 1 to show ribbons and the ligand.
preset apply int 1
Now the first serious problem. There is no command to make the morph. We need to use some Python code to do this step. That code is in morph.py and we can use the "open" command to run it.
open morph.py
How would you create this Python code? If you are a Python programmer you might study the Morph dialog code in your Chimera distribution chimera/share/Morph/gui.py. Otherwise you would write to the Chimera users mailing list chimera-users@cgl.ucsf.edu asking us to provide this bit of morphing code.
Hide the original unbound model (#1), and everything but the ligand of the bound state model (#0).
~modeldisplay #1 ~ribbon #0 show ligand
Show residues in the morph (model id #2) within 5 Angstroms of the ligand.
show #2 & ligand zr<5
The morph is in the unbound state and we want to show all residues in the bound state. We go to the last frame of the morph (bound state) using a bit of Python -- there is no equivalent Chimera command.
open lastframe.py
Now show the nearby residues with this frame.
show #2 & ligand zr<5
and return to the unbound state, the first frame of the morph.
open firstframe.py
Make the background color gray, the morph ribbon white, the ligand yellow.
set bg_color gray ribcolor white #2 modelcolor yellow ligand
repr bs ligand
There are no commands to enable silhouette edges and increase subdivision quality so we again resort to using bits of Python code.
open silhouette.py open quality.py
Now we name the 3 positions for the ligand flying into the binding site.
savepos closeview
Zoom out and move molecule to lower left corner.
scale 0.7 move x -20 move y -10 savepos wideview
Freeze the morph (#2) and move the ligand to the upper right corner and rotate it some.
~select 2 move x 60 move y 40 turn z 90 turn x 90 select 2 savepos farligand
Add a caption
2dlabel create title text "Binding maltotriose" xpos 0.3 ypos 0.92 color black
Now we play the sequence we wish to see in the movie.
Ligand flying in. Caption fades out.
reset wideview 50 reset closeview 25 2dlabel change title visibility hide frames 25
Morphing to the bound state requires some Python code.
open playmorph.py
Let's zoom in and show hydrogen bonds between the ligand and protein and rock the view.
scale 1.03 25 hbond intermodel true intramodel false color pink linewidth 5 roll y 0.5 50 roll y -0.5 50
Create a surface and color it white and orient vertically.
surface #2 color white,s #2 turn z 3 30
We can fade in the surface with a sequence of commands that give the percentage of transparency.
surftransparency 50 #2 surftransparency 25 #2 surftransparency 0 #2
Color aromatic residues blue.
color blue aromatic ring
Rock and zoom out.
rock y 3 150 scale 0.98 50
We put all these commands in a single file with a text editor. We insert some "wait" commands and the movie record/stop/encode commands to actually capture the movie.
Restart Chimera and try opening this command file to record the full movie.