Tom Goddard
November 8, 2016
NIH site-visit
This ChimeraX demonstration exploring nuclear pore architecture will highlight recent developments related to Technology Research and Development (TR&D) projects 1 and 2, and two driving biological projects (DBP 1 and 3) and a collaborative project (CSP 1) described in the grant proposal.
|
|
|
|
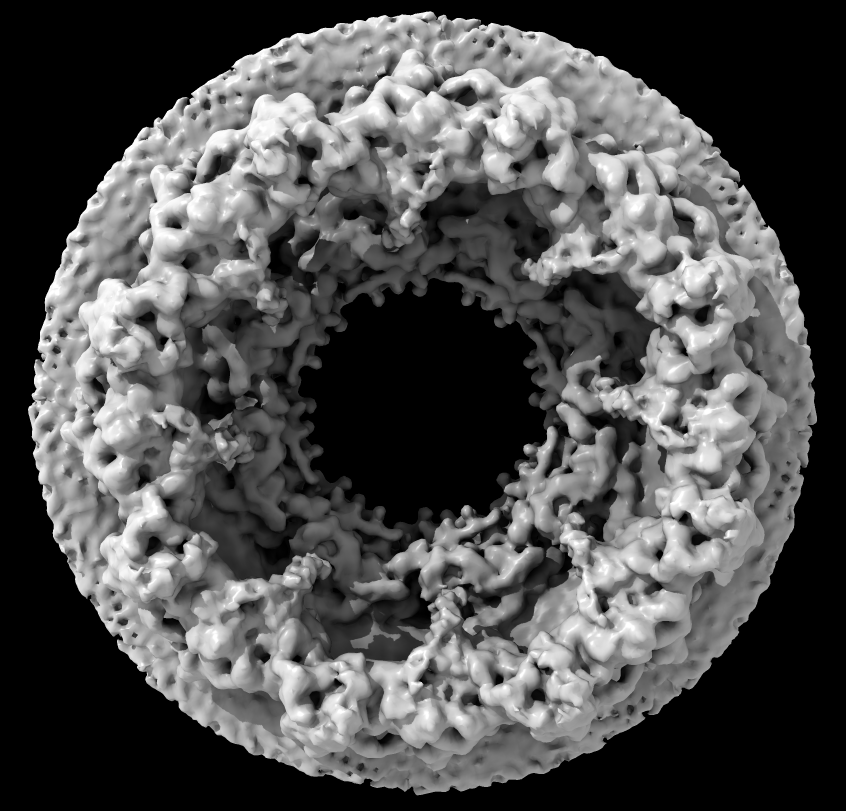
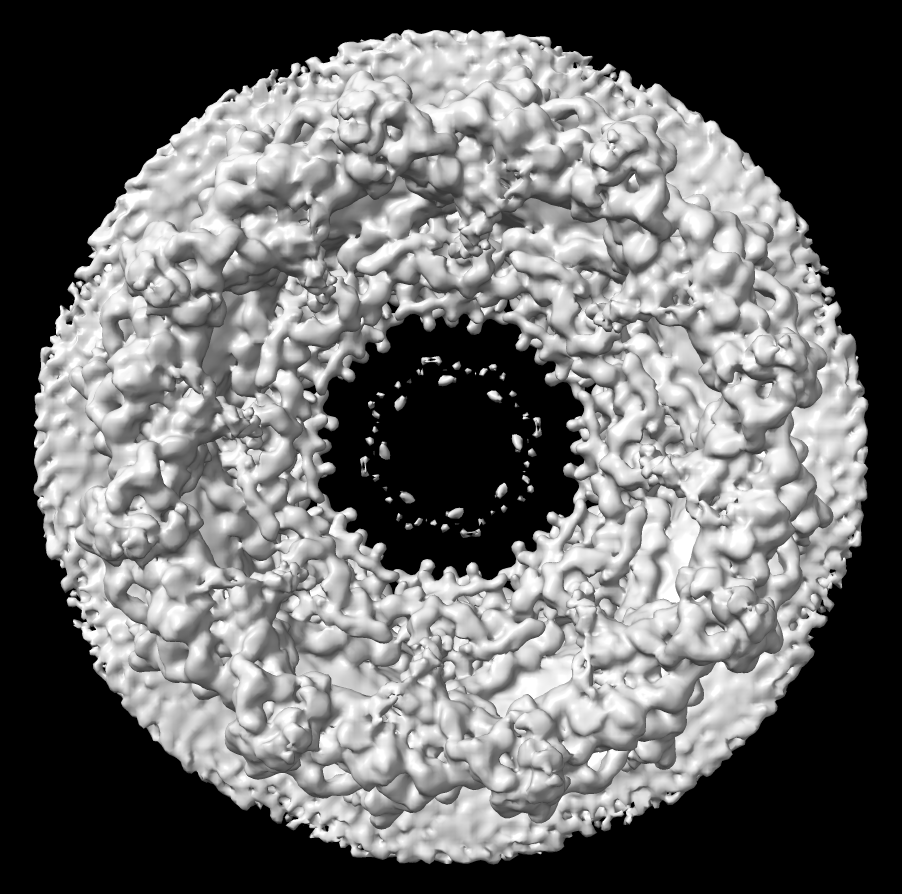
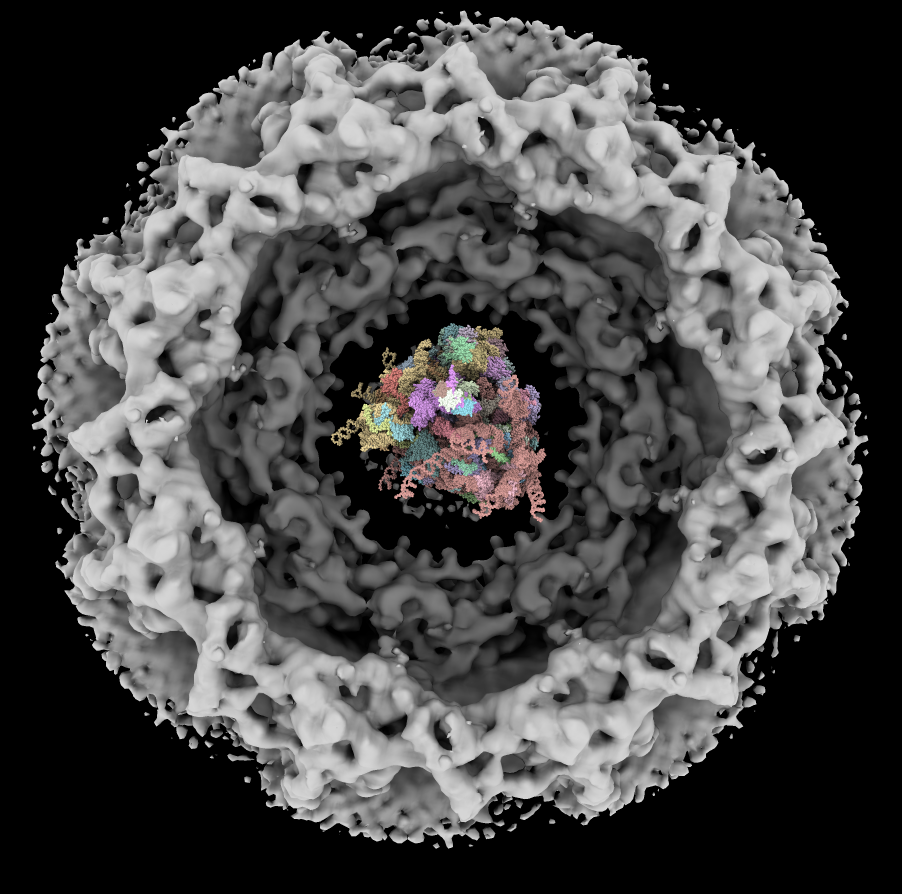
| Human nuclear pore EM, 23 Angstroms resolution (Beck lab). Ambient occlusion lighting shadows crevices. | Simple lighting used by current generation visualization programs, hard to perceive depth. | Ribosome size compared to nuclear pore. |
Nuclear pore atomic model
|
|
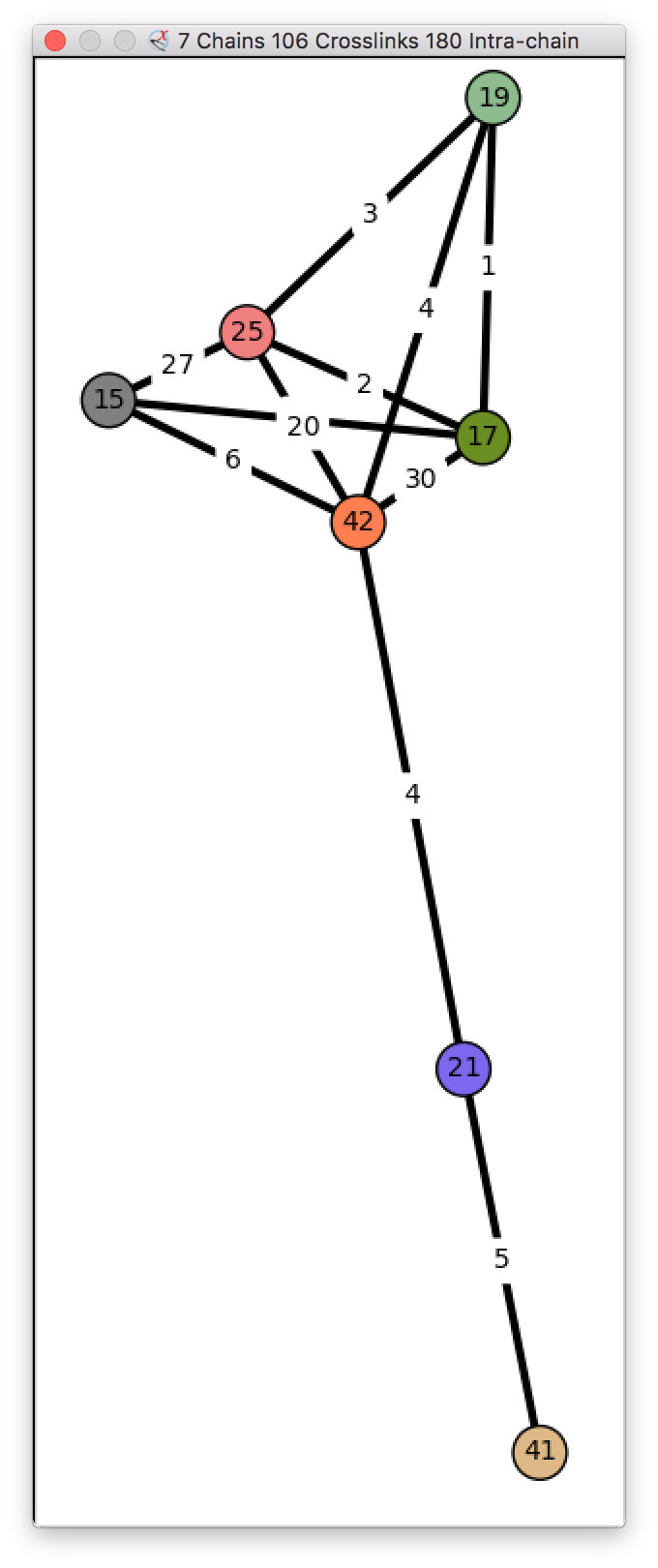
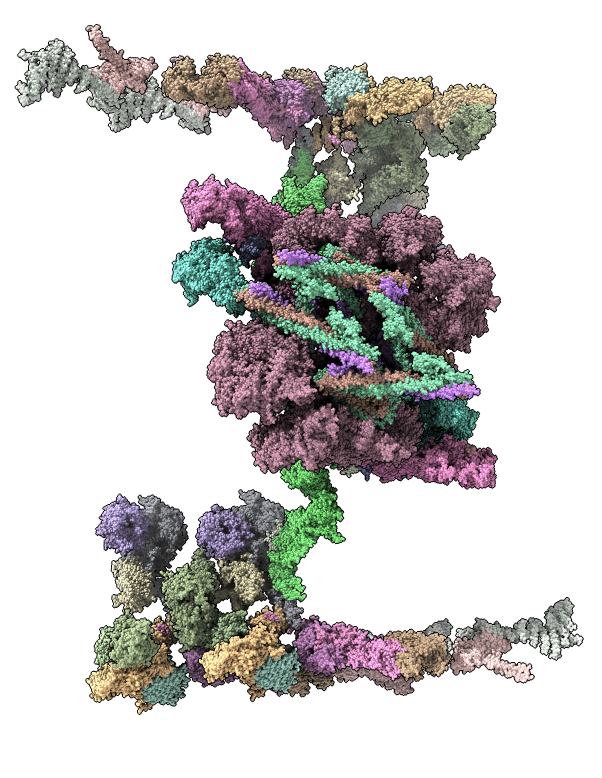
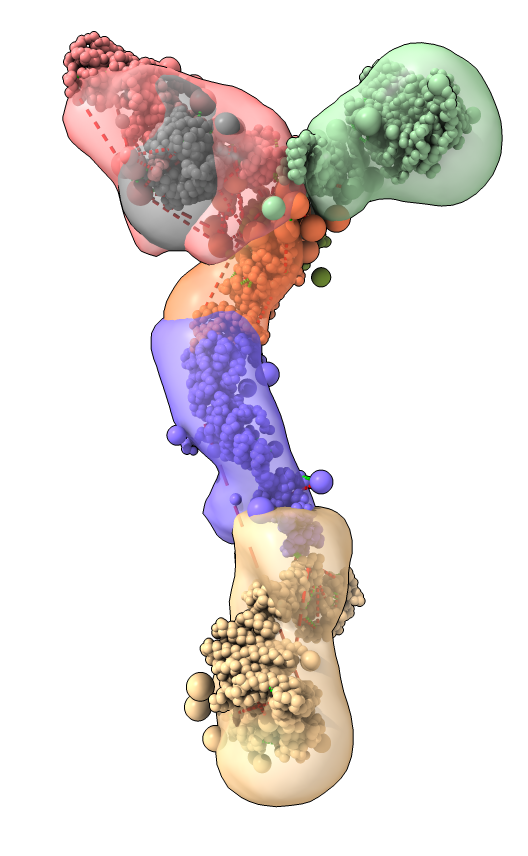
Integrative hybrid model of Y-complex
|
|
| 
| 
|
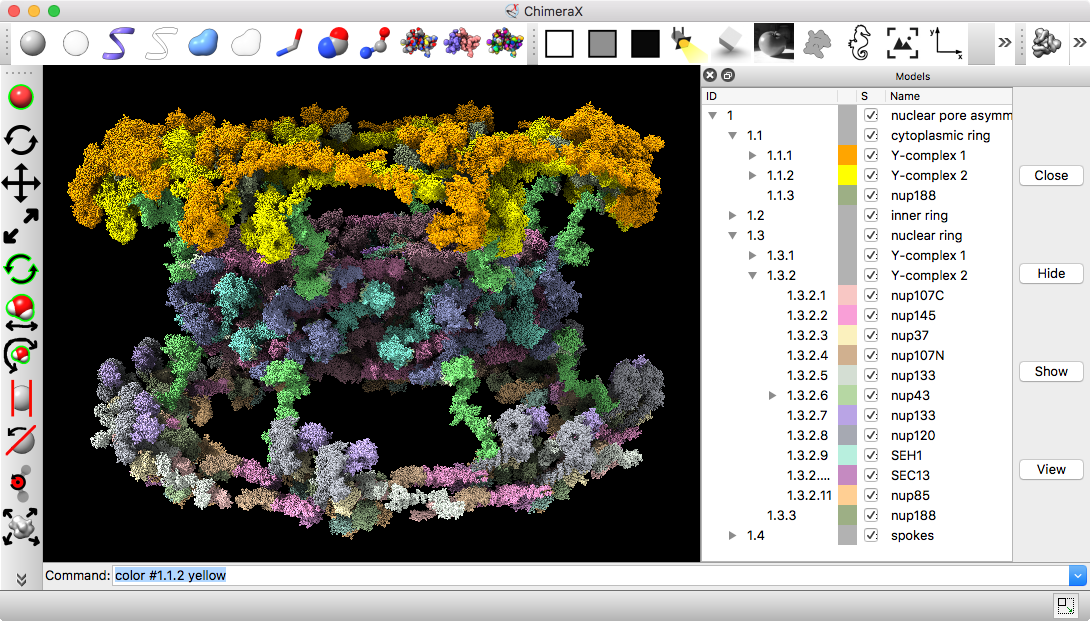
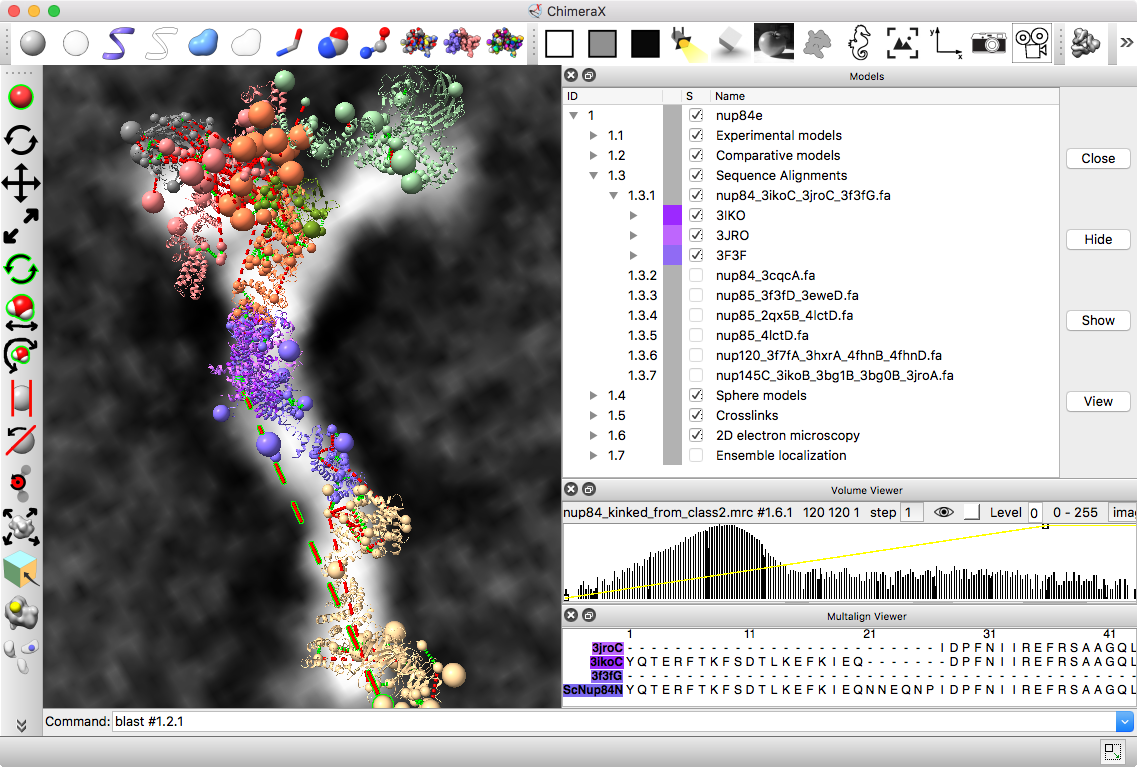
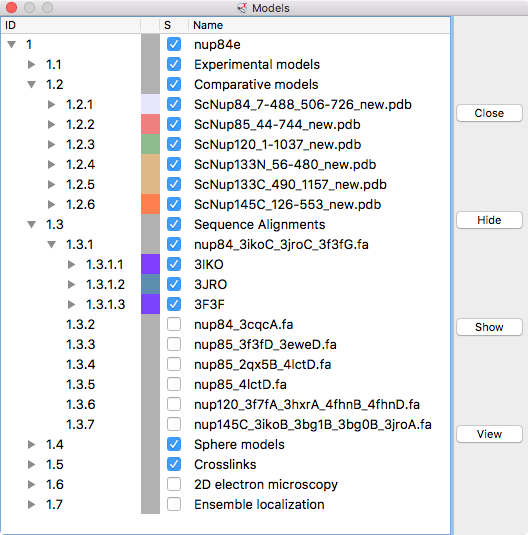
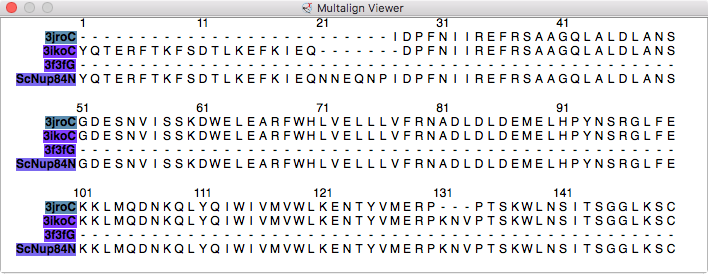
| Models from IHM file | Template sequence alignments for one protein. | Superimposed template models on comparative model. |
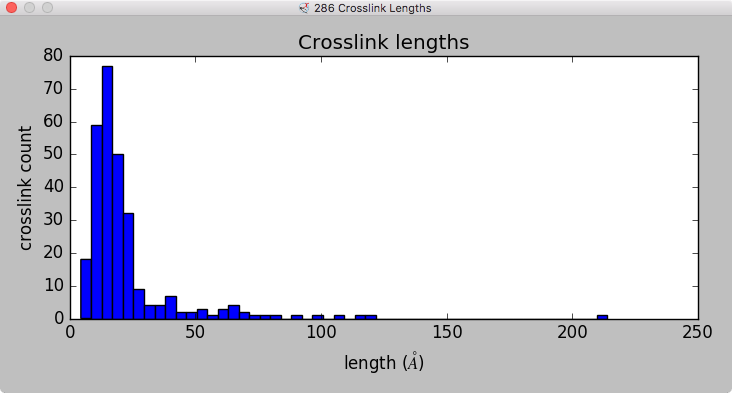
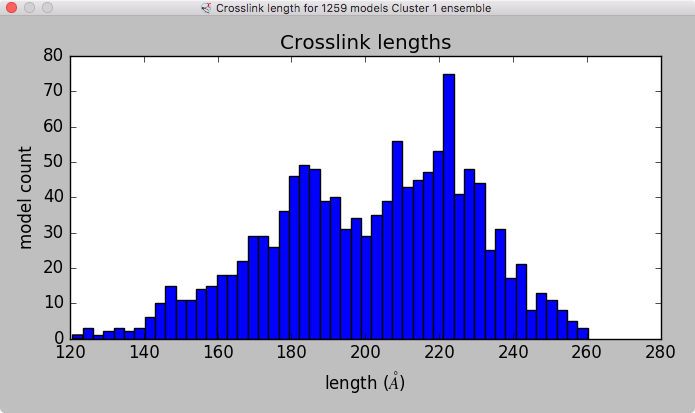
Model ensemble from sampling and scoring restraints
|
|
|
|
| 
| 
|
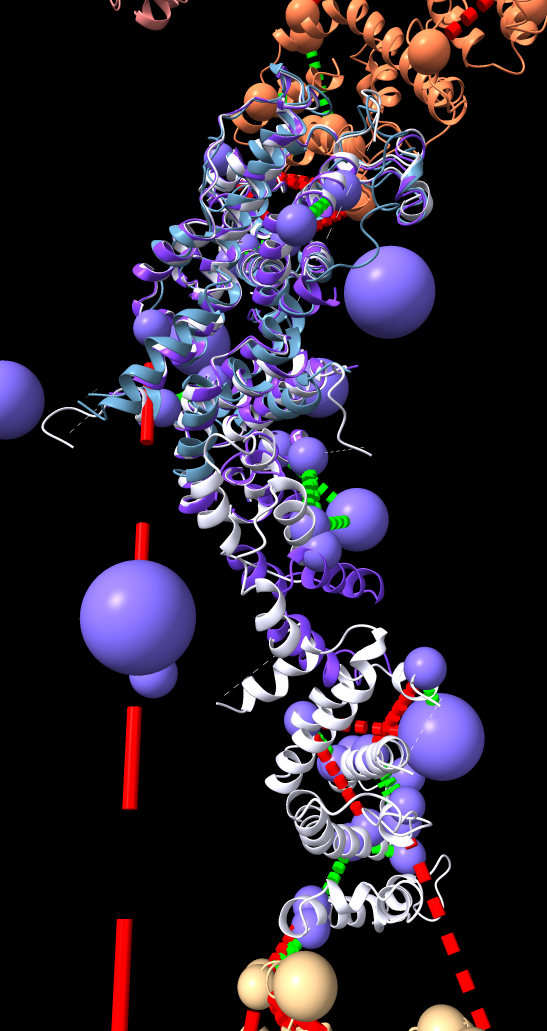
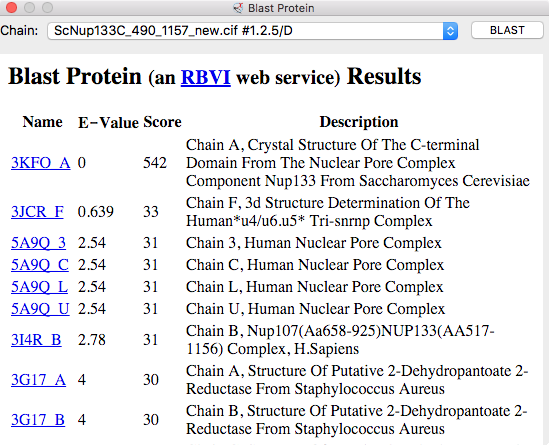
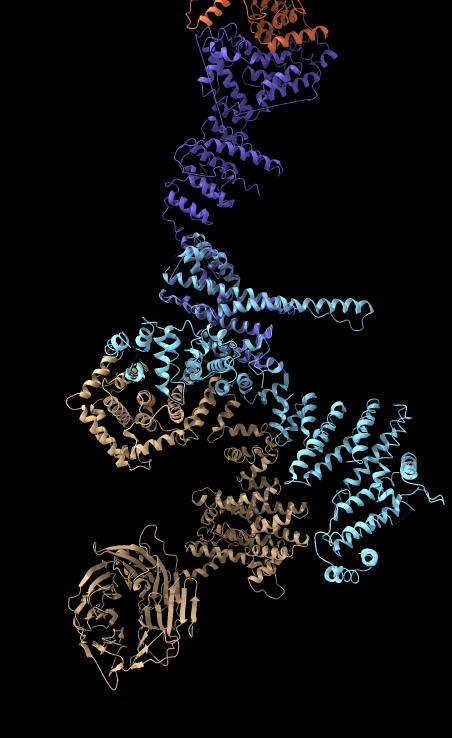
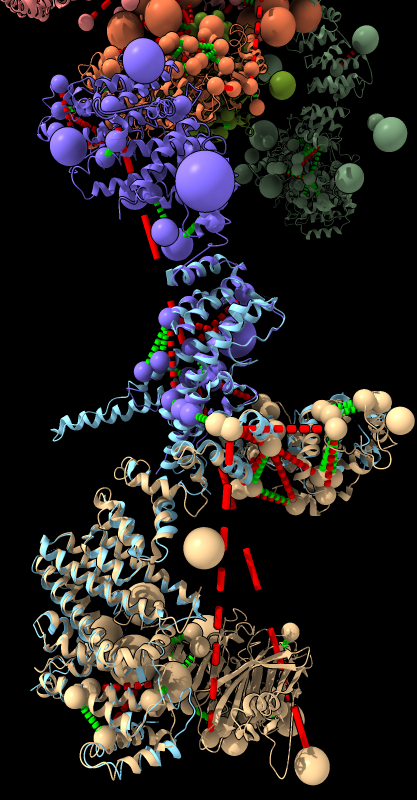
| BLAST PDB nup133 sequence | 3ikr aligned to nup84 (aka nup107) | 3ikr (light blue) bridging nup84/nup133 |