Structure Measurements
The Structure Measurements panel
has three sections shown as index cards:
Only one card is shown at a time, and clicking the tab for another
brings it to the front. Each section is listed as a tool
in the Structure Analysis category. Tools can be started in
several ways.
Save saves measurements to a file.
Close closes the measurements panel, and Help opens
this manual page in a browser window.
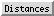
The Distances section of
Structure Measurements
is a table of distance monitors
(measurements that update if there are changes).
See also:
distance
There are several ways to start
Distances, a tool in the Structure Analysis category.
Distance monitors can be created in three ways:
- The Create button
creates a distance monitor between two atoms that have been
selected
(exactly two atoms must be selected).
- Picking one
atom and
Shift-double-picking
the second atom (that is, doubleclicking it with the button
assigned to picking)
elicits a button marked Show Distance.
Clicking it creates the distance monitor
and leaves the atoms in a
selected state.
Clicking elsewhere removes the button without creating a distance monitor
but leaves the atoms in a
selected state.
- The command
distance can also be used.
Atoms can be listed in a simple style consisting of residue
name, residue specifier (number.chain), and atom name, as shown in the figure,
or with command-line
specifiers (see the Adjust Torsions figure).
Atomspec display style in the
General preferences controls which style is used.
Clicking on a distance listing
allows it to be deleted with the Remove button.
If there is only one distance monitor, it is not necessary to click
on its listing before using Remove.
Clicking on a distance listing
selects the
corresponding distance monitor
pseudobond
and deselects any others; if the distance monitor is already
selected, it is deselected.
The Labels setting applies to all distance
monitors and can be switched among:
- None - no label
- ID - distance monitor ID number (the first column in the listing)
- Distance - the distance in angstroms
The Decimal places and Show Angstrom symbol settings
control how distances are reported in the table and
(when Labels is set to Distance) in labels.
The Decimal places can be changed by clicking or holding down
one of the arrows on either side of the value.
Of course, decimal places beyond those present in the input
coordinates are not very meaningful.
The lines drawn to represent distance monitors are
pseudobonds in a group named distance monitor.
Clicking Display options... brings up a list of
pseudobond attributes
for this group, allowing changes in properties
such as color, linewidth, and whether the lines are dashed.
When an individual
pseudobond
is selected,
its attributes
can be altered with the
Selection Inspector.
The Angles/Torsions section of
Structure Measurements
is a table of angle monitors (measurements that update if there are changes).
A "bond angle" is measured for three atoms and a "torsion angle"
for four atoms; however, it is not necessary for
the atoms to be contiguous or even bonded to one another.
Angles/Torsions reports changes but cannot be used to change the angles.
To modify torsions, use Adjust Torsions instead.
There are several ways to start
Angles/Torsions, a tool in the Structure Analysis category.
Angle monitors can be created in various ways:
- The Create button
creates an angle monitor for atoms that have been
selected (exactly
three or four atoms must be selected; a bond angle is measured when
three atoms are selected, a torsion angle when four atoms are selected).
- Picking
two atoms and then
Shift-double-picking
the third (that is, doubleclicking it with the button
assigned to picking)
elicits a button marked Measure Angle.
Clicking it creates a bond angle monitor and leaves the atoms in a
selected state.
Clicking elsewhere removes the button without creating a monitor
but leaves the atoms in a
selected state.
- Picking
three atoms and then
Shift-double-picking
the fourth (that is, doubleclicking it with the button
assigned to picking)
elicits a button marked Measure Torsion.
Clicking it creates a torsion angle monitor and leaves the atoms in a
selected state.
Clicking elsewhere removes the button without creating a monitor
but leaves the atoms in a
selected state.
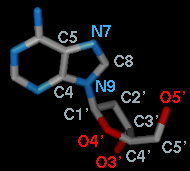
Atoms can be listed in a simple style consisting of
residue name, residue specifier (number.chain), and atom name
(see the Distances figure)
or with command-line
specifiers (see the Adjust Torsions figure).
Atomspec display style in the
General preferences controls which style is used.
Clicking on a listing allows it to be deleted with the Remove button.
If there is only one angle monitor, it is not necessary to click
on its listing before using Remove.
The Decimal places setting controls how angles are reported
in the table; the number of digits shown after the decimal
can be changed by clicking or holding down one of the arrows on
either side of the value.
The command angle
can also be used to measure bond angles and torsions; however, it yields
a static measurement rather than a continuously updating monitor.
The Adjust Torsions section of
Structure Measurements
is a table of active (rotatable) torsions.
See also: Rotamers,
swapaa
There are several ways to start
Adjust Torsions, a tool in the Structure Editing category.
Torsions can be activated in several different ways:
- The Activate button
activates a rotation around a bond that has been
picked
in the graphics window.
- Double-picking
a bond (that is, doubleclicking it with the button
assigned to picking)
elicits a menu with the option Rotate Bond.
Choosing this option activates the torsion,
opens Adjust Torsions (if it is not already open),
and leaves the bond in a
selected state.
Clicking elsewhere removes the menu
without activating a torsion but leaves the bond in a
selected state.
- The command
rotation or
brotation can also be used.
An error message will appear
if an attempt is made to activate a bond that is
terminal (no additional atoms attached to one or both ends),
within a ring, or already rotatable.
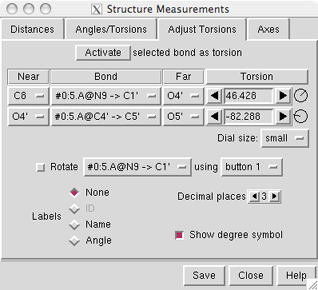
Atoms can be described with
command-line
specifiers, as shown in the figure, or in a
simple style consisting of residue
name, residue specifier (number.chain), and atom name
(see the Distances figure).
Atomspec display style in the
General preferences controls which style is used.
If the four atoms defining a torsion are called 1-2-3-4,
1 is the Near atom and 4 is the Far atom,
which will move when the bond is rotated.
The angle in degrees as defined by the current Near and Far
atoms is shown in the Torsion column.
Torsion can be toggled to Delta;
the reported value is then
the angle in degrees relative to the starting angle,
and there are no Near and Far columns.
 A bond can be rotated by entering a new angle value (and pressing return),
clicking the arrows flanking the angle value,
or manipulating the dial. The Dial size can be set to
small, medium, or large. Further, torsions
can be manipulated in the graphics window with the mouse.
This can be done by checking
Rotate [torsion] using [button]
and choosing the desired torsion and mouse button from the pulldown menus.
A bond can be rotated by entering a new angle value (and pressing return),
clicking the arrows flanking the angle value,
or manipulating the dial. The Dial size can be set to
small, medium, or large. Further, torsions
can be manipulated in the graphics window with the mouse.
This can be done by checking
Rotate [torsion] using [button]
and choosing the desired torsion and mouse button from the pulldown menus.
The Bond column contains a pulldown menu for each active rotation,
labeled with identifiers for atoms 2->3 (those flanking the rotatable bond):
- Revert - restore the original torsion angle
- Reverse - change which side of the torsion moves when the bond
is rotated (change the torsion definition from A-B-C-D to D-C-B-A)
- Deactivate
- deactivate the bond rotation and remove it from the table.
Note that the torsion will not automatically revert to its original value when
deactivated; Revert should be used before Deactivate to retain
the original torsional angle. Even if the same rotation is reactivated later,
Revert will not work to restore the angle, since the original value
has not been saved.
- Select - select
the rotatable bond and deselect any others;
when in the Torsion mode, also
select
the two flanking bonds (to show all four atoms defining the torsion).
When atom 2 is bonded to more than two atoms, there is more than one
possible Near atom, and alternatives (if any) are available
in a pulldown menu from the current Near atom name. Likewise,
when atom 3 is bonded to more than two atoms, there is more than one
possible Far atom, and alternatives (if any) are available
in a pulldown menu from the current Far atom name.
For the first torsion in the example (figures above), there are two choices
for the Near atom, C8 and C4, and two choices for
the Far atom, O4' and C2'. For the second torsion,
there are two choices for the Near atom, O4' and C3',
and only one possible Far atom, O5'. Of course, if
Reverse is used, the Near and Far choices are
interchanged, and in the Delta mode,
there are no Near and Far columns.
The Labels setting applies to all active torsions
and can be switched among:
- None - no label
- ID - torsion ID number (not yet implemented)
- Name - identifiers for atoms 2->3
(as shown in the Bond column)
- Angle - the angle in degrees
The Decimal places and Show degree symbol settings
control how angle values are displayed
when Labels is set to Angle.
The Decimal places can be changed by clicking or holding down
one of the arrows on either side of the value.
The Axes section of
Structure Measurements
is a table of axes defined for protein helices and/or other sets of atoms.
Axes are displayed in the graphics window as rods.
See also:
PipesAndPlanks
There are several ways to start
Axes, a tool in the Structure Analysis category.
Clicking Define axes... opens a dialog for specifying atom sets and
other axis parameters:
- Each helix in [models]
- define an axis for each peptide/protein helix in the molecule model(s)
chosen in the list. Peptide/protein helix assignments
are taken from the input structure file or generated with
ksdssp.
Only the backbone atoms N, CA, C are used to define the axes.
- Selected atoms (axis name [aname])
- define a single axis named aname using all
selected atoms.
- axis display parameters:
Clicking Apply (or OK, which also closes the dialog) calculates
the axes, adds them to the table, and generates the corresponding displays.
Axes are defined in a two-step process:
- the axis is anchored at the geometric center (centroid) of the atomic
coordinates and aligned with the principal component of the coordinates
- the orientation is adjusted to reduce the spread of atom-axis distances
The table lists axis Name and Length, and entries can be
sorted by either by clicking the column header.
Axes can be chosen with the left mouse button.
Ctrl-click toggles the status of an individual axis.
A block can be chosen by dragging, or by clicking its first (or last) line
and then Shift-clicking its last (or first) line.
Choosing two axes reports their crossing angle and closest distance
(treating the axes as infinitely long) below the table and in the
Reply Log.
Optionally, choosing axes also:
- selects axis - selects
each axis (currently nothing can be done to such a selection, but the axes
are highlighted in the graphics window)
- selects atoms - selects
the atoms used to define each axis
Clicking Distance measures distances between the chosen axes
and selected atoms;
results are sent to the
Reply Log.
If a single atom is selected, the distance to each chosen axis is reported;
if multiple atoms are selected, the minimum, maximum, and mean distances
are reported.
Deleting any of the atoms used to define an axis or closing their models
deletes the axis.
UCSF Computer Graphics Laboratory / March 2008