
AddH adds hydrogen atoms to molecule models. Chimera uses atom and residue names, or if these are not "standard," atomic coordinates, to determine connectivity and atom types; AddH then uses the atom types to determine the number of hydrogens to be added and their positions. The positions of pre-existing atoms are not changed, but any lone pairs and unidentifiable-element atoms are deleted.
 There are several ways to start
AddH, a tool in the Structure Editing category
(including using it via
Dock Prep).
AddH is also implemented as the command
addh.
There are several ways to start
AddH, a tool in the Structure Editing category
(including using it via
Dock Prep).
AddH is also implemented as the command
addh.
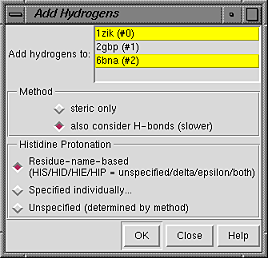
Models to add hydrogens to can be chosen from the list with the left mouse button. Ctrl-click toggles the status of an individual model. To choose a block of models without dragging, click on the first (or last) and then Shift-click on the last (or first) in the desired block.
Even when only one model is chosen, hydrogen placement can be affected by other models perceived to interact with the chosen model. Different submodels of the same model are not considered to interact.
The Method for adding hydrogens can be:
Clicking OK initiates hydrogen addition and dismisses the dialog, while Close merely dismisses the dialog. Help opens this manual page in a browser window.
If any atoms cannot be assigned a type, another dialog will appear. It is necessary to click on the line for each unassigned atom and then indicate its proper substituent geometry and number of substituents.
Bond lengths for X-H (X = C/N/O/S) are taken from the Amber parm99 parameters:
| X | atom types | X-H bond length (Å) |
|---|---|---|
| sp3 carbon | C3 | 1.0900 |
| sp2 carbon | C2,Car | 1.0800 |
| sp carbon | C1 | 1.0560 |
| nitrogen | N3+,N3,Npl,Ng+ | 1.0100 |
| sp3 oxygen | O3 (except water) |
0.9600 |
| sp3 oxygen | O3 (water) |
0.9572 |
| sulfur | S3 | 1.3360 |
The default VDW radii of carbon, nitrogen, oxygen, and sulfur atoms depend on whether hydrogen atoms are present. Therefore, the radii of some atoms will change when hydrogens are added.
When a more intensive approach is desired, the program Reduce (developed by the Richardson Laboratory) is a good alternative. Reduce places hydrogens to optimize local H-bonding networks and avoid steric overlaps, while flipping certain sidechains 180 degrees as deemed appropriate to fulfill these criteria. Asparagine and glutamine sidechains may be flipped to switch their terminal N and O atoms, and the imidazole ring of histidine may be flipped to switch N and C identities. The protonation state of histidine is adjusted based on the local environment.
Reduce is available free at http://kinemage.biochem.duke.edu:
J.M. Word, S.C. Lovell, J.S. Richardson, and D.C. Richardson, "Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation" J Mol Biol 285:1735 (1999).