


Tom Goddard
April 28, 2005
The atomic force microscope (AFM) scan of a crystal of satellite tobacco mosaic virus (STMV) shown below reveals details on individual particles. This detail may be noise, debris on the surface, afm scan artifacts, or damage to particles. Here we look at whether averaging particles can reveal the pentamers making up the virus capsid.
The AFM data comes from Alex McPherson's lab at UC Irvine.
|
|
|
|
|
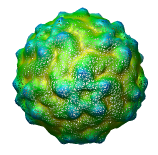
Figure 1: AFM data of crystalized virus. The scan is 256 by 256 data points (height measurements) and is nominally 400 nm on a side. It shows about 600 virus particles. Color coding is by height, lowest regions are red and highest are blue.


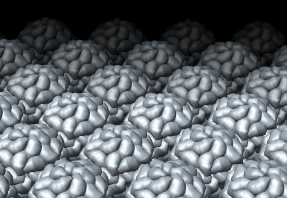
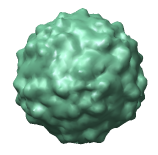
Figure 2: Capsid protein surfaces from atomic coordinates. A 1.8 angstrom crystal structure of satellite tobacco mosaic virus was determined in 1998, PDB model 1a34. A crystal plane (Miller indices 10onebar) is shown with similar packing as in the AFM data. I do not know if it is the same crystal plane as in the AFM data, or even if the crystals for the AFM and x-ray data have the same space group. This is just to see the icosahedral arrangement of capsid proteins. Each capsid protein is represented by a low resolution surface computed from atom density using 8 angstrom bins. Surfacing individual proteins exaggerates the boundaries between proteins.



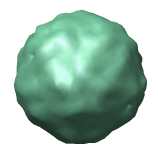
Figure 3: Virus particle surfaces from atomic coordinates. Here the crystal plane from PDB model 1a34 is shown with each virus particle being surfaced separately instead of each capsid protein surfaced separately as in figure 2. The divisions between proteins are not apparent. The particle surfaces are computed from atom density using 8 angstrom bins as explained in figure 4.
| 
| 
| 
| 
|

| 
| 
| 
| 
|
| 
| 
| 
| 
|
| 5 Angstroms | 10 A | 15 A | 20 A | 30 A |
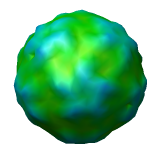
Figure 4: Virus particle surfaces at different resolutions from atomic cooordinates. Virus surfaces shown at different resolutions using crystal structure 1a34. The surfaces were calculated by placing all 240,000 atoms in a grid of bins of sizes 5, 10, 15, 20, or 30 angstroms and taking a contour surface of the atom density at 0.02 atoms per cubic angstrom. Also 2 iterations of smoothing were done moving each vertex 0.3 of the way towards the average position of neighboring vertices. This was to hide the triangles making up the surface. The top row shows the surface in a single color. The second row shows the surface radially colored (yellow at 80 angstroms radius, blue at 95 angstroms). The third row shows the surface mesh indicating the sampling. Notice that at 20 or 30 angstroms the icosahedral symmetry is not even apparent in the radially colored surface because of the coarseness of the sampling. The AFM data in figure 1 has sample spacing 15.6 angstroms.


Figure 5: Marking AFM particles. We interactively place a 10 by 10 grid of markers with the mouse using extensions to UCSF Chimera. Moving the 3 red markers at the corners stretches and skews the lattice. When it approximately aligns with the virus particles the lattice of markers can be extended to the whole surface. Get lattice parameters a = 18.85 nm, b = 15.47 nm, theta = 54.17 degrees. The 214 white spheres mark lattice cells that were used to compute an average particle, while the black spheres mark locations not included in the average. The markers to include were obtained by averaging 16 particles and then taking all particles that have RMSD within 1 nanometer of that average.

| 
| 
|

|
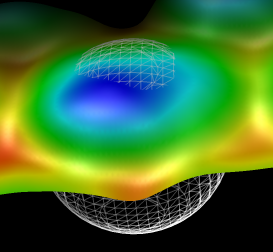
Figure 6: Average AFM particle. Average of 214 particles marked with white spheres in figure 5. Doesn't show any recognizable capsid structure. Color coding is by z height, red for low regions to blue for high regions covering a 4 nanometer range. Mesh sphere is 16 nm in diameter.

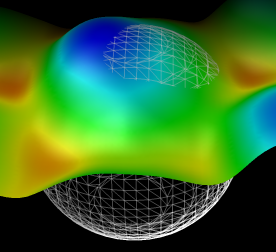
Figure 7: Residuals. The result of subtracting the average particle shape from the original AFM data at the 214 positions used for computing the average.
The resolution needed to see boundaries between closely packed proteins can be much higher than the size of the proteins in question if they pack to form a smooth surface. The STMV capsid protein is about 3 by 5 nm. The AFM data has 1.5 nm sample spacing with very probably lower resolution. Even with 1.5 nm resolution there is little hope of resolving capsid proteins no matter how many particles are averaged. It may be intersting to apply averaging to data that shows capsid structure in individual particles, such as AFM scans of Turnip Yellow Mosaic virus crystals or Paramecium Bursaria Chlorella virus trisymmetron plates. For enveloped viruses with heterogeneous capsid structure, averaging is not useful.
Data file. Data was from Aaron Greenwood, file STMV10221636.001 with header date Oct 22, 1998.
Plane fit. The AFM data file header indicates a plane fit was subtracted off by the acquisition software "Plane fit: -178 3406 525.5 2". I believe the slope of the plane in x was -178/65536 and in y 3406/65536. That's about 0.2 degrees tilt in x and 3 degrees tilt in y.

Crystal planes. The 1onebar0 crystal plane of 1a34 has slightly different packing than the 10onebar plane with a little larger gap between particles on the horizontal lines. Lattice parameters for the 10onebar 1a34 crystal plane are a = 19.18 nm, b = 16.44 nm, theta = 54.33 degrees, while the AFM data has a = 18.85 nm, b = 15.47 nm, theta = 54.17 degrees.
Particle size. Virus particle diameter measured from particle spacing along the close packing axis of the AFM scan is 15.47 nanometers. From PDB model 1a34 the spacing is 16.44 nanometers.
AFM scan is not flat. The crystal plane should be flat. Along the scan it is quite flat. Perpendicular to the scan direction it is not flat, exhibiting wavy variations of about 3 nanometers amplitude. The left image shows a side view of AFM data with scan lines in image plane, right image is side view with scan lines perpendicular to image plane. Both have height scaled by a factor of 5. White bar is a reference straight line and has thickness 2 nm (scaled thickness is 10 nm). The tilt of the left scan is the actual tilt of the data (exaggerated by factor of 5 scaling). It had a plane fit subtracted by the acquisition software but that does not make it level because the plane fit included the regions where the top crystal plane is missing and the large scan artifacts.


|
Fourier transform. The fourier transform of the AFM height data shows large components at all frequencies perpendicular to the scan lines with low frequencies parallel to scan lines (left edge of image in red). This suggests that each successive scan line has a sizable random height offset. This is very probably an artifact of the microscope. |

|
Surface rendering algorithm. The afm surface rendered in figure 1 was obtained using a cubic interpolation to double the number of data points in each dimension. That interpolated data is used to create a triangulated surface by dividing each grid cell into 4 triangles meeting at a center point. The center point is given height equal to the average height of the 4 grid cell corner heights. Simpler surface display schemes did not show as much detail and showed undesirable artifacts.

| 
| 
| 
|

| 
| 
| 
|
| right diagonal | left diagonal | isotropic | cubic interpolation |
Four surface triangulations are shown in four columns. The images in the first row show the surface appearance, and in the images in the second row show the triangle mesh for a single particle. The first two methods use only the original data values for surface height. Notice that the diagonal orientation is faintly visible in the surface images. That is bad. The third column places an extra point at the center of each grid cell with height equal to the average of the 4 surrounding data values. That surface uses twice the number of data points and triangles. The fourth column is the method I used for images. Twice as many data points are obtained along each axis by doing a cubic interpolation, and a center point is placed in each cell of the resulting grid to avoid anisotropic artifacts. This last method shows the single pixel detail from the original data best but uses 8 times the number of data points and triangles as the simplest method.

| 
|
| 2 rounds of cubic interpolation | |
Two rounds of cubic interpolation shows a little better quality but requires 4 million triangles to render the 256 by 256 data set. That large a number of triangles can only be rotated at 1 frame per second with my moderately fast graphics card (Quadro4 500 GoGL).